

























General Neurology
Use of focused ultrasound in neurologic disorders
Jan. 13, 2025
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
Primary eye care providers are usually the first ones to encounter unusual retinopathies but confuse them with other entities, such as optic neuritis. Neuro-ophthalmologists are the primary referral points, although referral to general neurology usually occurs if such conditions are accompanied by headaches or dizziness. Patients with retinopathies and choroidopathies, if unrecognized, are usually referred by their primary eye care providers to neuro-ophthalmologists, general neurologists, or uveitis subspecialists (for choroidal conditions), although all groups of subspecialists see all these types of patients. Occasionally, a patient may be referred to a multiple sclerosis specialist if the primary eye care provider misdiagnoses a retinal condition as an optic neuritis. This article addresses the more unusual forms of retinopathies and choroidopathies that might prove challenging to diagnose. Such uncommon pathologies include Usher syndrome, systemic lupus erythematosus, polypoidal choroidal vasculopathy, and punctate inner choroidopathy, among others.
|
• Occult retinopathies and choroidopathies can be misdiagnosed as other conditions, such as optic neuropathies. | |
|
• Clinical examination, including a thorough medical history, is essential for diagnosis. | |
|
• Using ancillary tests such as ocular coherence tomography (OCT) has helped in the early diagnosis of conditions that are not readily apparent with routine ophthalmoscopy, eliminating the need for advanced neuroimaging. | |
|
• Prompt diagnosis and treatment of retinopathies and choroidopathies is essential to avoiding further complications and permanent blindness. | |
|
• Recognizing the underlying systemic disease may prove critical to treat the eye disorder (eg, systemic lupus erythematosus or HIV). |
These two conditions present in patients as deafness and progressive night blindness as a result of retinitis pigmentosa. Retinitis pigmentosa is frequently recognized by pigmentary changes in the retinal pigment epithelium. Such changes, however, are sometimes not apparent with routine ophthalmoscopy. Both conditions are inherited in an autosomal recessive manner. Of these two, Refsum is by far the rarer. Usher syndrome, on the other hand, occurs with a relatively high prevalence in persons of Acadian descent where common ancestry is a factor (ie, Louisiana).
Definition. Hereditary motor and sensory neuropathy type IV is a rare autosomal recessive disease caused by defective alpha oxidation of phytanic acid. The defective enzyme is phytanoyl-coenzyme A hydroxylase, which normally catalyzes the second step in the breakdown of phytanic to pristanic acid (217; 80). This disease can cause a retinal degeneration indistinguishable from retinitis pigmentosa.
Pathophysiology. The retinal degeneration seems related to the excessive deposition of phytanic acid in ocular tissue. Studies have shown complete loss of photoreceptors, thinning of the inner nuclear layer, and reduction in the number of ganglion cells of the retina (43).
Epidemiology. Retinitis pigmentosa affects 50,000 to 100,000 people in the United States and an estimated 1.5 million people worldwide. According to some authors, 4% to 5% of patients with retinitis pigmentosa may have Refsum disease (209).
Clinical manifestations. The onset is usually late in childhood or adolescence (43), and it follows a slowly progressive course (216). The cardinal neurologic manifestations of the disease include a demyelinating neuropathy, pes cavus, cerebellar ataxia, sensorineuronal deafness, anosmia, and cranial nerve involvement. Nyctalopia and visual failure secondary to retinitis pigmentosa often precede the neurologic symptoms; visual fields are constricted; and cataracts and photophobia exist. Impairment of pupillary light responses with miosis possibly caused by high lipids is part of a generalized dysautonomy. Also, nystagmus is reported in 25% of patients (130).
Diagnostic workup. Retinal exam shows pigmentary changes resembling “bony spicule” or “salt and pepper” (76).
Electroretinography (ERG) may show reduction or complete absence of rod and cone responses. Retinitis pigmentosa with constricted visual fields and night blindness are characteristic features and commonly the first clinical symptoms.
Nerve biopsies showing “onion bulb” and targetoid inclusions have been described in Schwann cells, which have a similar appearance on electron microscopy. Nerve conduction studies show delayed responses. CSF protein levels are usually elevated. Plasma levels of phytanic acid are elevated (more than 19 mmol/l or up to 800 mmol/l) (72).
Prognosis. Over the years, visual field constriction develops gradually until, finally, only tubular vision remains.
In addition to retinitis pigmentosa, visual failure in patients with Refsum disease may also be aggravated by optic atrophy, cataract, and vitreous opacities (223).
Management. Phytanic acid dietary restriction reduces plasma and tissue levels. Fish, beef, lamb, and dairy products should be avoided. Average intake should be reduced to 10 to 20 mg/day (202).
Significant advances have been made in the treatment of this disease. A 14-year-old girl diagnosed with Refsum was treated with a phytanic acid-poor diet and extracorporeal lipid apheresis over a 30-month period, after which she was treated with diet only (117). Membrane filtration and heparin-induced extracorporeal low-density lipoprotein precipitation apheresis were well tolerated. During a 5-year period, blood phytanic acid levels decreased to a noncritical range. She remained free of ophthalmologic and neurologic progression for a total observation period of 12 years.
Roca-Saavedra and colleagues offer an extensive review of the literature on phytanic acid content in foods, the management of the phytanic acid content during food production, and the biochemical mechanisms of phytanic acid metabolism (175).
Definition. Usher syndrome is an autosomal recessive disorder characterized by congenital sensorineural hearing deficit of varying severity with or without balance disorders and progressive visual loss secondary to retinitis pigmentosa.
Etiology. There are a variety of genetic forms of Usher syndrome named in Table 1.
Usher form | Chromosomal location | Protein |
USH1A | 14q32(66) | - |
Pathogenesis and pathophysiology. In pathological studies, findings include degeneration of the organ of Corti, cochlear ganglion atrophy, anomalies of cilia on photoreceptor cells and cilia of olfactory receptor neurons, slow sperm motility and an abnormal structure of the sperm, bronchiectasias, and reduced nasal mucociliary clearance (205).
In the mouse retina, myosin VIIA is detected in the outer layer of the optic cup, which will give rise to the pigment epithelium cells. This protein, although present in both the pigment epithelium cells and the photoreceptor cells in all species tested for Usher syndrome, contains molecular defect, resulting in cell death (21).
A mutation of USH2A 2299delG has been detected at the heterozygous state in 4% of patients affected by a recessive form of isolated retinitis pigmentosa.
Epidemiology. This syndrome is the most frequent cause (50% to 66%) of deafness accompanied by blindness. About 18% of retinitis pigmentosa cases and 3% to 6% of congenital deafness cases have been associated with this disease (26). Its prevalence is between 1/16,000 and 1/50,000, based on studies of Scandinavian, Colombian, British, and American populations; for individuals between the ages of 30 and 49, the prevalence approaches 1/10,000 (96). In the United States, the prevalence of Usher syndrome has been estimated to be 4.4 per 100,000. In relation to the genotypes of Usher syndrome in the United States and Northern Europe, it is reported that 33% cases are USH1, and 44% cases are USH2 (87). In Colombia, however, 70% of the Usher syndrome cases have been diagnosed as USH1 (206).
Clinical presentation. Researchers have described three types of Usher syndrome: type I, type II, and type III.
Usher syndrome type I. Usher syndrome type I is the most severe type--characterized by severe to profound congenital sensorineural deafness, constant vestibular dysfunction (manifested clinically as a delay in motor development and ataxia), prepubertal onset retinitis pigmentosa that appears as loss of night vision and a restriction of the visual field during childhood, and, eventually, as a visual acuity loss that rapidly progresses to blindness (192; 137).
Usher syndrome type II. Individuals with Usher syndrome type II have less severe deafness (a mild hearing loss for low frequency sounds and a severe hearing loss for high frequency sounds) with absence of vestibular dysfunction. The loss of night vision develops around puberty with progression of the visual field impairment (220).
Usher syndrome type III. Individuals with Usher syndrome type III are born with normal hearing but develop retinitis pigmentosa with late progressive hearing loss and occasional presence of vestibular dysfunction.
Diagnostic workup.
Usher syndrome type I. Anomalies of electroretinography can be detected as soon as 2 to 3 years of age, thus, permitting early diagnosis of the disease. Later, fundus anomalies develop, which consist of pigment deposition-like bone spicules around the mid-periphery of the retina, thereafter extending inward and outward, and a narrowing of blood vessels.
Usher syndrome type II. Patients usually have funduscopic anomalies beyond childhood (111).
|
• The Kay pictures test and the Sheridan-Gardiner test are widely used tests for preverbal children and can be applied to deaf children as well. | |
|
• Fundus examination for identification of retinal or optic nerve abnormality is only performed by a specialist for patients at any age. | |
|
• Electroretinography will reveal functional deficit of the outer retinal layers even before ophthalmoscopic signs are present. Infants are a concern because the contact lens electroretinography recording requires sedation or anesthesia in young patients and presents a risk of corneal abrasion. For this reason, skin electroretinography has been introduced and is widely used, using suitable stimulus conditions and signal averaging without the need for sedation. Although the exact sensitivity and specificity of electroretinography in identifying Usher syndrome in very young children remains to be determined, electroretinography is widely accepted as the best available method for screening deaf children (233). | |
|
• Balance tests. Conventional vestibular testing methods are usually not well tolerated by young children. However, methods such as the video or charge-coupled device camera inspection of eye movements (video nystagmoscopy undertaken while children are rotated on the parent’s lap) appear to be promising methods of assessing vestibular function (118). In cases where the child does not cooperate for rotary chair testing even on his or her parent’s lap, alternative testing such as the rapid doll’s eye test can be used (85). |
Prevention. In the study by Mets and colleagues, the earliest diagnosis was at 6 months of age using electroretinography in a child with severe to profound deafness who subsequently received a cochlear implant (2 years old) (153). Early diagnosis and timely intervention are essential if the development of communication skills is to be optimized.
Prognosis and complications. According to Grondahl, the prognosis for visual function is favorable for the autosomal dominant type (82). Some patients showed good visual function until 30 years of age, but few had useful visual function after the age of 50 years.
Management. Although research on visual implants is improving (236), this technology is currently not applicable. In the absence of any effective treatment, gene therapy approaches have emerged for the treatment of retinal degeneration in general. This is possible by accessing the retina with local application of therapeutic vectors such as adenovirus, adeno-associated viruses, and lentiviral vectors for the delivery of genes into the retinal cells in vitro and in vivo (38). For a current review of the pathogenesis, molecular diagnosis, and therapeutic approaches to Usher syndrome, the reader is referred to (166).
Definition. Retinopathies present in association with embolic disease represent a major risk for cardiovascular events.
Etiology. Retinal ischemia is often caused by emboli arising from the cardiac chambers or the common carotid artery bifurcation (the latter are often composed of cholesterol).
Pathogenesis and pathophysiology. Venous stasis retinopathy and ocular ischemic syndrome are associated with severe hypoperfusion of the eye and usually reflect severe carotid occlusive disease.
Retinal emboli may originate from ulcerations of the atheromatous carotid artery, from mural thrombi in the carotid artery, or from cardiac valvular structures after a myocardial infarction.
Fibrinogen is a major factor in the development of vascular disease. Studies have shown that elevated fibrinogen levels may contribute to atherosclerosis by stimulating smooth muscle migration and proliferation, platelet aggregation, and increased blood viscosity.
Epidemiology. Retinal arteriolar emboli are associated with an increased risk of cerebrovascular disease mortality; 15% of those who experience retinal arteriolar emboli die within a year and 54% die within 7 years.
After cardiac bypass surgery retinal emboli are found twice as often as compared to baseline eye exam before cardiac bypass. Association of cardiovascular disease and retinal emboli increases with smoking, hypertension (OR 2.2), and diabetes mellitus (OR 0.7). The Beaver Dam Eye Study estimated that there are 1.2 million people in the United States aged 43 to 86 years with retinal emboli in at least one eye, about 450,000 of whom are 75 to 86 years of age (115).
Clinical presentation. Patients with carotid artery disease may present with ipsilateral ocular symptoms and signs that can herald a devastating stroke. Patients with retinal emboli may complain of transient monocular visual loss, and in such cases branch or central retinal artery occlusion are the most common final diagnosis. Eighteen percent of the asymptomatic patients with retinal emboli have carotid stenosis of more than 50% to 75%.
Stereoscopic fundus photographs of the disc and macula may show retinal emboli or other lesions similar to those in hypertensive retinopathy:
|
• Retinal aneurysm |
Diagnostic workup. The source of the emboli must be determined. For a carotid artery source, consider using carotid duplex, computed tomographic arteriography (CTA), or magnetic resonance arteriography (MRA). For aortic arch and cardiac sources, use transthoracic ECHO, transesophageal ECHO (TEE), or 24-hour Holter.
Prevention. Retinal artery occlusion and retinal arteriolar emboli are shown to be associated with hypertension, higher systolic blood pressure, carotid artery plaque, and increased plasma fibrinogen levels. Retinal vein occlusion is additionally associated with increased diastolic blood pressure, body mass index, arteriovenous nicking, and focal arteriolar narrowing. Retinal arteriolar emboli are additionally associated with prevalent coronary heart disease, increased plasma lipoprotein (a) levels, and cigarette smoking.
Management. Patients can be managed with control of arterial hypertension, dyslipidemia, and blood glucose. Patients should be instructed to use aspirin and stop smoking. In a meta-analysis of the literature, the efficacy of statin treatment for venous thromboembolism was explored (124). Statin treatment was compared with a placebo or no treatment. The authors concluded that observational and intervention studies suggest a beneficial effect of statin as an adjunct to anticoagulant therapy in venous thromboembolism prevention and that rosuvastatin performed better than other statins. There was no evidence of an effect of statin use on pulmonary embolisms (124). For a current review that focuses on aspects of retinal and optic nerve ischemia that may be encountered by neurologists, the reader is referred to (22).
Definition. Polypoidal choroidal vasculopathy was first described as a posterior uveal bleeding syndrome with multiple recurrent retinal pigment epithelial detachments (231). The choroidal circulation is involved, and the characteristic lesion is in the inner choroidal vascular network of the vessels, ending as an aneurysmal bulge and visible as a red, spheroid, polyp-like structure (234). Polypoidal choroidal vasculopathy is now thought to be a distinct form of type 1 (under the retinal pigmented epithelium) choroidal neovascularization. The diagnosis of polypoidal choroidal vasculopathy is best made using indocyanine green angiography because it permits visualization of the choroidal vasculature (05).
Histopathology. Analysis of a polypoidal lesion from a 76-year-old Japanese man showed degeneration of the walls of large, dilated venules (159). Degenerated small arterioles and capillaries with thickened basement membrane were also identified. Massive exudation of fibrin and plasma, along with hyalinization of choroidal vessels, were prominent features in all specimens of polypoidal choroidal vasculopathy lesions (158). The role of VEGF in the pathogenesis of polypoidal choroidal vasculopathy is not clear. Studies have shown that polypoidal choroidal vasculopathy is resistant to anti-VEGF therapy, suggesting that the development of polypoidal choroidal vasculopathy is not VEGF-related (77; 39).
Etiology. Taken together, genetic studies suggest that polypoidal choroidal vasculopathy may share common mechanisms with neovascular age-related macular degeneration, as well as have specific mechanisms of its own (135). No serum biomarker for polypoidal choroidal vasculopathy has yet been identified. Because the primary lesion in polypoidal choroidal vasculopathy is believed to be in the choroidal vasculature, the atherosclerotic aging processes in large vessels likely contribute to the development of polypoidal choroidal vasculopathy as well (203).
Pathogenesis and pathophysiology. Microscopic findings describe abnormal vessels sometimes associated with lymphocytic infiltration located under the retinal pigment epithelium (126).
Polypoidal choroidal vasculopathy has been associated with age-related macular degeneration in more than three case reports in the past 10 years. The common link between multifocal choroiditis, burned-out chorioretinal inflammatory lesions, and polypoidal choroidal vasculopathy is the possible stimulation of embryonic rest by inflammation, which leads to vascular proliferation and the lesions of polypoidal choroidal vasculopathy.
Epidemiology. A predisposition for African-American and Asian females has been described (35). Polypoidal choroidal vasculopathy was first thought to be rare and to affect mostly black women. It is still thought to occur most often in pigmented individuals (blacks and Asians) but is now recognized in white patients as well (232). The age of diagnosis can range from the 20s to the 80s (125), but polypoidal choroidal vasculopathy is most commonly diagnosed between the ages of 60 and 70 (101).
Clinical presentation. This is a bilateral disease and may present with varied symptoms and eye findings such as (188):
|
• Vitreous hemorrhage | |
|
• Minimal fibrous scarring | |
|
• Absence of drusen | |
|
• Disc edema with peripapillary hemorrhages | |
|
• Myopia | |
|
• Signs of intraocular inflammation | |
|
• Subretinal bleeding |
The clinical manifestations of polypoidal choroidal vasculopathy were reviewed, and these are most often seen in the posterior segment. Variably sized serous and hemorrhagic detachments of the neurosensory retina and pigment epithelium around the optic nerve or in the macula are seen ophthalmoscopically. A typical presentation for a patient who is symptomatic for less than 3 months is extensive subretinal exudation and bleeding with minimal cystic changes in the retina, leaving the patient with good visual acuity (101).
Diagnostic workup. Fundus photography along with high-resolution optical coherence tomography, fluorescein angiogram, and indocyanine green angiography are the mainstays of the diagnostic work-up (101). ICG is still the gold standard for ocular imaging; its longer wavelengths penetrate the retinal pigment epithelium and allow improved imaging of the choroidal vessels, making for easy identification of the polypoidal choroidal vasculopathy (136). The appearance of the lesion often depends on its localization in the posterior pole (48):
|
• Juxtapapillary lesions |
- radial arching pattern | |
|
• Lesions limited to the macula |
- oval distribution pattern | |
|
• Juxtapapillary and macular lesions |
- irregular latticework |
Size of the lesions tends to be around 0.5 disc diameter and has intense uniform fluorescence on the ICG (147).
Differential diagnosis. Differential diagnosis can be classified in three major groups:
|
(1) Vascular malformation | |
|
The last three entities are excluded because of the staining of the optic nerve during ICG. Others are excluded by evidence of choroidal or scleral thickening, and the finding of fluid in the sub-Tenon space on ultrasound examination (198) | |
|
(2) Peculiar type of choroidal tumor |
Prognosis and complications. Good visual prognosis is expected for those lesions that do not involve the macula.
Management. A conservative approach is recommended unless lesions are associated with persistent or progressive exudative change that threatens central vision.
Laser photocoagulation results in resolution of the disease (127). Reduced vision may occur from photocoagulation of vessels extending to the center of the fovea. Alternative methods in addition to subfoveal laser are vitrectomy and submacular removal of the polypoid structure (99).
Several other methods of treatment for polypoidal choroidal vasculopathy have been reported. The principal therapies are laser photocoagulation, photodynamic therapy, anti-vasogenic drugs, and combinations of these. The efficacy of the anti-vasogenic drugs has been brought into question as they have shown little or no therapeutic effect in this condition, suggesting that polypoidal choroidal vasculopathy may not depend on VEGF in the same way as neovascular age-related macular degeneration (77). In another study, the authors reached a different conclusion, that, “Intravitreal anti-VEGF injection monotherapy may be a valuable therapeutic option in treating eyes with sub macular hemorrhage associated with polypoidal choroidal vasculopathy” (40). The on-going problems with monotherapy have come under critical review in the treatment of wet age-related macular degeneration and it is likely that the same will occur in the treatment of polypoidal choroidal vasculopathy. In an excellent review of all forms of ocular neovascularization, Campochiaro elucidates the molecular pathogenesis of subretinal and retinal neovascularization and then discusses targets for interrupting this process. Anti-vascular endothelial growth factor, anti-platelet derived growth factor, and agents that will target hypoxia inducible factor-1 might be combined to produce a much more robust therapy against this devastating disease process than presently exists. He summarizes by saying, “While substantial progress has been made, the future looks even brighter for patients with retinal and choroidal vascular diseases” (34). A single case report demonstrated significant improvement in a patient with subretinal fluid secondary to peripapillary polypoidal choroidal vasculopathy when treated with oral eplerenone (109).
|
Choroidal neovascularization |
Polypoidal choroidal vasculopathy | |
|
Lesion appearance |
Smaller caliber vessels, grayish |
Evident red to orange lesions |
|
Discoloration of the overlying retina and easily detected in clinical examination |
Ending with secular polyp evident with slit lamp (09) | |
|
Subretinal thickening not manifested |
Subretinal thickening manifested by polyp-like structures | |
|
Histopathologic findings |
Diminishing thickness of the choroid |
No demonstration of any of these characteristics (179) |
|
ICG |
Diffuse late-staining plaque |
Prominent vascular network (195) |
|
Clinical location |
Fibrotic and disciform scar |
Pigment epithelium detachment (213) |
|
Prognosis |
Severe macular changes leading to loss of vision |
Not necessarily macular change and better outcome after treatment |
One final note on the differentiation between choroidal neovascularization (eg, exudative age-related macular degeneration) and polypoidal choroidal vasculopathy is in order. In a genetic study of 568 unrelated Chinese individuals (156 exudative age-related macular degeneration patients, 164 polypoidal choroidal vasculopathy patients, and 248 controls), meta-analysis of the genetic data showed a strong and consistent association of the ARMS2/HTRA1 locus with both exudative age-related macular degeneration and polypoidal choroidal vasculopathy, suggesting that the two disorders share, at least partially, similar molecular mechanisms (132).
Definition. Shaken baby syndrome is a form of childhood physical abuse with few outward signs of physical injury. It is seen with whiplash-like injury from forcible shaking while being grabbed by the shoulders or torso, resulting in subarachnoid hemorrhage. Shaken baby syndrome is often accompanied by signs of strangulation, nutritional deprivation, and environmental neglect (65).
Etiology. Shaken baby syndrome is the result of whiplash-like injury and rotational forces affecting axons and vessels.
Pathogenesis and pathophysiology. The severe force, high velocity, acceleration, and deceleration imposed on the unsupported head and neck result in severe intracranial and intraocular damage. Biomechanical studies of these injuries suggest that the magnitude of forces are 50 times greater than when the head is forcefully struck against a surface, propagating from the inside out, with no protection of the neural tissue of the head. The magnitude of these forces causes rupture of cerebral and ocular vessels within the subarachnoid space and can produce optic nerve sheath hemorrhage, retinal breaks, detachments, and dialysis. The layers of the retina can also be split, causing retinoschisis (167).
Epidemiology. Child abuse is defined as endangerment of a child at the hands of a parent or guardian. The incidence of child abuse in the United States is reported as affecting about one million children annually. And the incidence of shaken baby syndrome is 4000 cases per year. Ten percent of traumatic injuries in children are nonaccidental; of these, 10% to 20% have fatal injuries. Eye injury occurs in more than 40% of all child abuse cases and in up to 95% of shaken baby syndrome cases. Demographics show that victims at risk came from families of low socioeconomic status, minority races, and low levels of education.
Clinical presentation. The affected child is usually as old as 3 years of age, and the presentation often is with signs of strangulation, nutritional deprivation, and environmental neglect. Neurologic symptomatology may be, thus, relevant to cerebral edema with lethargy, vomiting, seizures, apnea, bradycardia, hypothermia, and bulging fontanels. There may be nutritional aspects and electrolyte and fluid imbalance, skin bruising, and fractures. Subarachnoid hemorrhage, subdural hemorrhage, and intraparenchymal bleeding can be excluded with a brain scan (84). Ocular findings are characterized by retinal hemorrhages that are often severe and proportional to the extent of the injury. Diagnosis is sure when retinal hemorrhage coexists with subdural hemorrhage and cerebral edema. Ninety-five percent of the babies have ocular findings; 90% of the findings are bilateral, but they can be asymmetrical. The anterior segment is normal, and pupillary reactivity may be sluggish with decreased visual responsiveness (173).
Deep extensive posterior pole retinal hemorrhages can involve all layers of the retina. A severe vitreous hemorrhage signals poor prognosis (196). A choroidal rupture is indicative of direct injury; or in the absence of blunt impact, it signifies severe trauma, and the child is unlikely to survive.
The following images are from a patient with shaken baby syndrome:
Retinal folds. Retinal folds usually occur in the perimacular area. Pathogenesis of this is not clear; some studies suggest traction between the internal limiting membrane and vitreous.
Retinal dialysis. In a patient whose head suffers an impact with a solid object, the retinas often are large and quadrantic. This is evident a few months after the trauma.
Retinoschisis. Retinoschisis induces an absolute scotoma and occurs at the cleavage plane formed within the retina, completely disconnecting the inner layer from the outer layer. Retinoschisis frequently originates from the periphery and extends to the posterior pole, affecting central vision. The traction force created by the schisis conforms an ongoing injury to the vitreous, deforming it and reformatting it almost immediately.
Retinal holes. Retinal holes are rarely present but can progress to retinal detachment when the subretinal fluid escapes the subretinal space (13).
On ERG, schisis is indicated by selective extinguished B waves. Retinal detachment reveals a normal or proportionate attenuated signal.
Differential diagnosis. Other causes of retinal hemorrhage in infants are:
• Traumatic birth |
For a current review of the diagnostic difficulties associated with shaken baby syndrome, especially where death has occurred, the reader is referred to (33). These difficulties have been expanded upon (139).
Prognosis and complications. Retinal folds usually tend to resolve by themselves and do not affect vision in the long term unless the fovea is affected. But retinal folds do have an association with severe brain damage and cortical vision loss (180).
Management. On presentation, the child should be assessed in the emergency room for any life-threatening injuries. After stabilized, the patient should undergo an extensive eye examination. Eyelid, anterior segment, and ruptured globe injuries may require anesthesia. Most intraretinal, subretinal, and preretinal hemorrhages clear spontaneously within 4 weeks after injury. Schisis, choroidal rupture, retinal folds, retinal detachment, and optic nerve trauma may be repaired surgically. Retinal detachment repair can be done by creating a barrier using laser photocoagulation around the detachment area (114). Paiva and colleagues summarized the issues related to clinical presentation, diagnosis, risk factors, and interventions in shaken baby syndrome (162).
Definition. Ocular siderosis is a complication induced by a chemical reaction to an iron intraocular foreign body (57).
Etiology. A retained iron intraocular foreign body may cause toxic effects, such as cataract or retinal damage, which can develop even if the object is removed. If the foreign body is small, a patient can remain asymptomatic for months before siderosis occurs (97).
Pathogenesis and pathophysiology. The retinal damage surrounding the foreign body object is related mostly to the impact of the iron intraocular foreign body and subsequent retinal damage.
Epidemiology. Siderosis may occur 18 days to 8 years after ocular injury.
Clinical presentation. If the iron intraocular foreign body is large, it can be associated with major disruption of the ocular structures. When small, the foreign body can be hidden by hemorrhages, cataracts, or hyphema. In cases of siderosis, anisocoria and abnormal pupillary reaction is present along with heterochromia. Corneal deposits are usually seen (rusted color), and cataract optic atrophy with arteriolar narrowing is seen in end-stage disease. The patient presents with a large scotoma corresponding to the pigmentary changes caused by the foreign body (97).
Diagnostic workup. The clinician should first exclude that there is an open globe and then proceed to look for the presence of the iron intraocular foreign body (193). Direct examination of the eye should be done promptly as cataract or hemorrhage may quickly obstruct the view. ERG will help detect ocular siderosis. The affected eye will have reduced “a” and “b” waves for both photopic and scotopic responses. The unaffected eye will gradually develop ERG abnormalities in full-blown cases of siderosis (100).
Prevention. Eye protection should be used when working with metallic objects.
Prognosis and complications. Complications depend on the content of iron in the object and the ocular structures affected in siderotic process (168). The effects are also dependent of the size and location of the foreign body. The visual field defect is permanent in the area of pigment degeneration (122).
Management. The severity of the injury determines the treatment. The main issue is removal of the iron intraocular foreign body using specific surgical techniques, which include the use of lasers and magnets (191).
Definition. Sclopetaria is a nonpenetrating contusion-type of injury of the eyeball characterized by rupture of the choroid and retina with hemorrhages caused by a missile (18).
Etiology. Sclopetaria is created when a missile, a BB for example, does not penetrate the eye but glances off the sclera and lodges adjacent to the globe in the orbit.
Pathogenesis and pathophysiology. The contusion causes two areas of injury, direct and indirect. Direct injury occurs where the missile strikes the choroid and the retina, producing fibriotic scarring of these structures. The indirect injury affects the macular area as a result of shock waves propagated through the vitreous, producing scarring (155).
Clinical presentation. The initial findings of macular edema and retinal hemorrhages are followed by atrophic scaring and hyperpigmentation.
Diagnostic workup. The history, clinical findings, and ocular ultrasound are crucial (226).
Prognosis and complications. Because the injury involves the macula, vision is seriously affected. Chronic hypotony is frequent. Common complications are retinal hemorrhages, retinal detachment, and angle recession (165).
Management. Sclopetaria is managed with monitoring of the ocular complication and with early intervention (146).
Definition. Central retinal vein occlusion can present with disc edema accompanied by dilation and tortuosity of the retinal veins and macular edema of varying degrees (140).
Etiology. The causes of central retinal vein occlusion can be classified into three categories: (1) external compression of the vein, (2) vascular disease or vasculitis, (3) thrombosis.
Pathogenesis and pathophysiology. Central retinal vein occlusion can be divided in nonischemic and ischemic types (169). There is an association of central retinal vein occlusion with:
|
• Open-angle glaucoma |
Epidemiology. Venous occlusive disease is the most common disease seen in clinical practice. Its incidence is 2:1000 in patients older than 40 years of age and 5:1000 in patients older than 65 years of age.
Clinical presentation and diagnostic workup. Visual acuity ranges from hand motion to 20/20. In the case of an ischemic process, the common visual acuity is 20/100, and patients with a nonischemic process tend to be less severe unless there is macular edema (88). In ischemic central retinal vein occlusion, some studies have found a relative afferent pupillary defect (RAPD) greater than 1.2 log; in nonischemic patients the RAPD is usually absent. Visual fields may show peripheral constriction, and central scotomas are more common in ischemic central retinal vein occlusion cases with severe macular edema.
Fundus findings are typically seen with cotton-wool spots; the incidence of 10 or more is indicative of an increased risk of rubeosis. Retinal hemorrhages, tortuous veins, and disc edema tend to disappear entirely after months. In some cases, microaneurysms can be seen with persistent macular edema and pigment irregularity.
Differential diagnosis. The diagnosis is clinical. The parameters that need to be assessed in the evaluation of the patient are visual acuity, degree of afferent pupillary defect, ophthalmoscopic findings, fluorescein angiography, and electroretinographic findings. Fluorescein angiography characteristically shows changes in vascular caliber and prolongation of filling after injection (185). Electroretinogram shows reduced B-wave amplitudes and prolonged B-wave implicit time.
Prevention. It is important to identify patients at risk with comorbidities, such as arterial hypertension, diabetes mellitus, hypercholesterolemia, previous CVA, and heart disease.
Prognosis and complications. When the disease is mild and there is little vascular dilation and few retinal hemorrhages, the prognosis is good. On the other hand, the condition that presents with multiple hemorrhages, cotton-wool spots, macular edema, and retinal edema is likely to develop rubeosis iridis and neovascular glaucoma, and the patient may become visually impaired.
Complications seen in central retinal vein occlusion are often macular edema and retinal hemorrhage with neovascularization (200).
Management. There is a growing trend away from laser photocoagulation in combination with steroids, and toward laser in combination with anti-VGEF agents or anti-VGEF therapy alone.
A survey of the literature assessed the effect and safety of using single intravitreal bevacizumab versus intravitreal triamcinolone acetonide and grid laser photocoagulation, or a combination of intravitreal bevacizumab and intravitreal triamcinolone acetonide for treatment of retinal vein occlusion-associated macular edema, by exploring its effects on visual acuity and central macular thickness. The authors concluded that intravitreal bevacizumab is effective in treating patients with retinal vein occlusion-associated macular edema; Furthermore, intravitreal bevacizumab seems to be safer than intravitreal triamcinolone acetonide because of the intraocular pressure increase often associated with the injection of steroids (138).
A meta-analysis of the literature compared anti-VEGF therapy with corticosteroid and laser therapy for macular edema secondary to retinal vein occlusion. It concluded that the anti-VEGF agents improved branch central vein occlusions and reduced central retinal thickness more effectively and longer than corticosteroid/laser treatment in eyes with retinal vein occlusions. Additionally, eyes treated with anti-VEGF agents had significantly lower intraocular pressures than eyes treated with corticosteroids/laser 3 and 6 months after initiating therapy (170).
Definition. Retinal vasculitis is a sight-threatening inflammatory disease characteristically affecting the retinal venules and, more rarely, arterioles commonly found in association with a systemic disease or in conjunction with other features of intraocular inflammation (PSII) (02).
For a current review of this condition “intended to establish a common understanding of the term, retinal vasculitis,” the reader is referred to “Retinal vasculitis” (176).
Etiology. The known causes and associations with retinal vasculitis are (01):
|
• Behçet disease |
Pathogenesis and pathophysiology. Although the mechanism of the disease is yet to be defined, there is increasing evidence of immune dysregulation and cellular processes involving various agents such as autoantigens like Retinal s-antigen (S-Ag) and interphotoreceptor retinoid-binding protein (IRBP), the active role of CD4 activation in the antigen presentation and immune mechanisms, and the role of the vascular endothelium and molecules such as selectins and integrins (98; 131). There have been reports of multiple sclerosis-related retinal vasculitis (64) and systemic lupus erythematosus vasculitis (98).
Epidemiology. The estimated prevalence of uveitis in the United States is approximately 204 per 100,000, with an estimated incidence in Western Europe varying from 7 to 19 per 100 000 per year. Incidence is increased in female patients younger than 40 years of age (222).
Clinical presentation. The clinical presentation is variable. If the problem remains in the periphery of the retinal vessels, the patient may be asymptomatic. In the majority of the cases, there is some degree of painless, blurry vision added to the underlying systemic symptomatology. Vision loss due to macular edema secondary to venous occlusion and leakage is seen in 60% of the patients with retinal vasculitis. There may also be “sheathing or cuffing” of the veins in the periphery, which can be segmental or confluent.
Vasculitis may result in leakage or occlusion of the lumen, which leads to retinal swelling, exudation, and edema with intraretinal hemorrhage, cotton-wool spots, and optic nerve head edema. And discussed in other retinopathies, nonperfusion and ischemia lead to neovascularization and vitreous hemorrhage that can conclude in retinal tear, detachment, or glaucoma (112).
Diagnostic workup. Fluorescein angiography is key in determining extent and severity of the disease, allowing us to see an immediate staining of the affected vessel wall and the surrounding tissues, even if there are minimal inflammatory changes that are not clinically significant. Capillary leakage, seen in 78% of isolated retinal vasculitis, is more common than ischemic macular edema. It is readily identifiable on angiography with typical petaloid pattern. New imaging techniques in retinal vasculitis have emerged. These include ultra-wide-field fluorescein angiography and optical coherence tomography angiography (151). For an excellent, easy to read, well-illustrated article on multimodal images in vasculitis, the reader is referred to Agarwal and associates (04).
Initial evaluation of a patient with no past medical history of any of the associated diseases should include erythrocyte sedimentation rate, C-reactive protein, complete blood count with differential, fluorescent treponemal antibody absorption test, Lyme and toxoplasmosis titers, chest x-ray, angiotensin-converting enzyme, antinuclear antibody, rheumatoid factor, basic chemistry panel, urinalysis, human leukocyte antigen typing, human immunodeficiency virus testing, and purified protein derivative with anergy panel (172).
If an infectious disease is suspected, serological testing should be performed or vitreous biopsy with polymerase chain reaction for cytomegalovirus and herpes simplex virus types 1 and 2, human T-cell lymphotrophic virus type 1 (HTLV-1), Toxocara, Brucella, Candida, and Leptospira species titers. If a systemic etiology is suspected and more detailed evaluation is warranted, then antineutrophil cytoplasmic antibody, complement levels, C3, C4, sinus x-rays, and anti-dsDNA antibodies can be ordered. If a neurologic disorder is suspected, then magnetic resonance imaging, lumbar puncture, and a neurology consult should also be considered.
Prognosis and complications. Patients with an underlying infectious disease will typically respond to antimicrobial agents. Corticosteroids may be necessary if residual retinal vasculitis remains. For patients with systemic disease, corticosteroids are the mainstay of therapy. Side effects are not uncommon, and systemic preparations should not be used for long-term therapy.
Management. The use of corticosteroids at doses greater than 10 mg per day in conjunction with other immunosuppressive agents with cyclosporine, azathioprine, or an alkylating agent is used to decrease the intraocular inflammation and to facilitate visual recovery and prevent long-term vision loss due to ocular sequelae (218).
An additional note about Behçet uveitis: Behçet disease is a multisystem immune-mediated vasculitic disorder afflicting many systems; ocular complications can be devastating for the patient and their quality of life. Eye involvement affects 60% to 80% of patients with Behçet disease and is characterized in its more severe form by posterior or panuveitis, including occlusive retinal vasculitis. For a review of current and future treatments of Behçet disease the reader is referred to Mesquida and colleagues (152). For a current review of the ocular manifestations of connective tissue diseases and systemic vasculitides, the reader is referred to (42).
Definition. Central retinal artery occlusion causes widespread retinal ischemia, with marked visual loss that is usually permanent in nature. Central retinal artery occlusion is a form of acute ischemic stroke. Analogous to cerebral ischemic stroke, central retinal artery occlusion is associated with a risk of recurrent vascular events.
Etiology. The occlusion may be due to an embolism, atherosclerotic changes, inflammatory endarteritis, or angiospasm. It is most often caused by emboli originating from atheromatous plaques in the carotid arteries or from a cardioembolic source. In some cases, the occlusion can be nonthrombotic due to infection or factor deficiencies (229).
Pathogenesis and pathophysiology. Ischemia begins after cessation of blood flow, which results in damage of the neural tissue and necrosis of the retina. Three mechanisms cause cellular damage in this process: (1) the lack of oxygen that alters all homeostatic mechanisms, (2) the ischemia of the neurons causing release of neurotoxins that allow entry of fluid and electrolytes into the cell, (3) acidosis causing peroxidation and formation of toxic free radicals. Besides the damage caused by the lack of flow, reperfusion injury is also known to cause damage. Reperfusion injury is caused by overstimulation by excitatory amino acids, the polysaturated fatty acids that are very susceptible to destruction by the free radicals. Vasoconstriction is caused by several factors that motivate the continuation of ischemia in the tissue (141). The most important determinant of retinal damage and final visual outcome is the duration of occlusion of the central retinal artery.
Epidemiology. The age- and sex-adjusted incidence of central retinal artery occlusion is 1.9 per 100,000 person-years in the United States. The incidence rises to 10.1 per 100,000 person-years in those over 80 years of age. They are seen more commonly in the elderly, and they are more prevalent in males. There is an association of cardiovascular conditions and risk factors with the presence of retinal arterial occlusion: carotid artery plaques and stenosis 75%, hypertension 67% to 78%, diabetes 25%, tobacco smoking and history of coronary bypass (181). Central retinal artery occlusion is most strongly associated with an ipsilateral internal carotid artery stenosis. In the European Assessment Group for Lysis in the Eye (EAGLE) study, 77 of 84 patients had a comprehensive workup for potential pathogeneses; 31 (40%) had over 70% carotid artery stenosis.
Nonarteritic retinal occlusion is most commonly due to a thrombus or an embolus that is visible only in 20% of the patients with retinal artery occlusion (49).
Clinical presentation. The classical presentation is with sudden painless vision loss. Relative afferent pupillary defect (RAPD) is present. Clinically, after 1 or 2 hours of progression, there is usually a cherry-red spot in the fundus as a result of the lack of arterial flow and a bright red coloration at the thinnest part of the retina; narrowing and irregularity in the arteries is also seen as is an irregularity in the caliber of the retinal veins and segmental opacification of the retina with cotton-wool spots, intraretinal hemorrhages, white-centered hemorrhages, retinal capillary microaneurysms, and telangiectasias (11).
Diagnostic workup. The diagnostic workup should be tailored to the age of the patient and the accompanying systemic neurologic symptoms. Patients older than 50 years of age likely will have atherosclerotic disease, and emboli from the heart or carotid should be sought with the appropriate test. Patients older than 65 years of age can be suspected as having giant cell arteritis, and Westergren sedimentation rates test should be ordered. Blood work can help determine the etiology if other testing is negative. A coagulation panel, antiphospholipid antibody levels, serum protein electrophoresis, and lipid panel may be helpful (186).
Differential diagnosis. Retinal detachment, retinal vein occlusion, and vitreous hemorrhage should be considered.
Prognosis and complications. Visual loss is usually permanent. Effort should be made for early diagnosis and intervention.
Management. Reliable systems to rapidly identify and treat patients with central retinal artery occlusion are being developed and are critical to both making treatment widely available and to ensuring adequate enrollment in clinical trials. The use of existing stroke code systems ensures rapid and reproducible evaluation of risk factors for hemorrhage. Intravenous tPA is an evidence-based therapy for acute ischemic stroke. It improves long-term functional outcomes in patients presenting within 4.5 hours of time last known with no evidence of intracranial or systemic hemorrhage. Since the 1960s, intravenous thrombolytic agents have been used empirically to treat central retinal artery occlusion, and tPA is currently administered in 5.8% of patients admitted with central retinal artery occlusion in the United States. In a patient-level meta-analysis of observational studies, Schrag and colleagues found that patients with acute central retinal artery occlusion treated with any lytic drug exhibited a 50% rate of clinical recovery when treated within 4.5 hours of onset (182). Conservative treatment includes ocular massage and paracentesis. Medical management with pentoxifylline, a vasodilator, might improve retinal blood, which may improve retinal blood flow following retinal arterial occlusion. Hyperbaric oxygen has been suggested as a way to preserve the retina in an oxygenated state, usually within 72 hours of the initial presentation. An invasive treatment includes catheterization of the proximal ophthalmic artery, usually through the femoral artery, with infusion of thrombolytic agents (178).
Definition. Vigabatrin is an antiepileptic drug used in management of complex partial, secondary generalized seizures and infantile spasms, with mechanisms based on the increased concentration of the g-aminobutyric acid that irreversibly binds to GABA transaminase (45).
Etiology. Vigabatrin inhibits the GABA transaminase enzyme, leading to increased GABA levels in cells in the retina (91).
Pathogenesis and pathophysiology. g-Aminobutyric acid is an inhibitory neurotransmitter and phototransductor from the retinal photoreceptor cells. When administered systemically, vigabatrin has been shown to cross the blood-retinal barrier and can be detected throughout the retina through immunocytochemical techniques. Pathologic studies have suggested that the visual complexity of vigabatrin toxicity is due to the loss of peripheral central cone-mediated vision (154; 16).
Clinical presentation. Bilateral-specific and symmetrical peripheral constriction in the visual field apparently occurs in 30% to 50% of the patients; the result is decreased visual acuity, color vision, contrast sensitivity, and central short-wavelength automated perimetry (51; 41).
Diagnostic workup. Examination of the visual field is the best way to detect serious changes in retinal function. ERG can be abnormal even if the visual field is normal. Photopic changes in ERG in 25% of children treated over a 6-week period with vigabatrin show early loss of oscillatory potentials, which is suggestive of cone, and in some cases rod, dysfunction. Other reports show B-wave abnormalities in adult patients on long-term vigabatrin therapy, indicating a toxic effect on the inner retinal layers (59; 121).
Prognosis and complications. Large doses of vigabatrin can cause abnormal ERG potentials that correlate significantly with the severity of the visual field loss. Discontinuing the vigabatrin treatment does not change either ERG potentials or visual field loss (86; 106; 79).
Management. There is no specific treatment other than discontinuing the drug.
Definition. Chloroquine and hydroxychloroquine are given for treatment of rheumatoid arthritis, systemic or discoid lupus erythematosus, and other connective tissue disorders.
Etiology. Studies have shown that the main cause for chloroquine or hydroxychloroquine toxicity are doses greater than 3.5 mg/kg (chloroquine) and 6.5 mg/kg (hydroxychloroquine) per day for more than 10 years plus renal insufficiency.
Pathogenesis and pathophysiology. Hydroxychloroquine has an affinity for pigmented tissue in the retina, binding with melanin-producing free radicals and toxins. Pathological studies have found the principal metabolite of hydroxychloroquine in ocular structures. The hydroxychloroquine effect is classified as pre-maculopathy and true maculopathy (211).
Epidemiology. In the United States, there is no definite report of the incidence of toxicity, but it is related to the dose and duration of treatment. Some authors have suggested an incidence of 10% in patients taking 250 mg of chloroquine per day, and 3% to 4% in patients taking 400 mg of hydroxychloroquine per day (214).
The risk of toxicity increases to 1% after 5 to 7 years of use. A cumulative dosage of more than 1000 g of hydroxychloroquine carries a significant risk of retinopathy (230; 144).
Clinical presentation. At presentation the patient may be asymptomatic, but usually the main complaint is paracentral scotoma. Patients also complain of decreased vision, glare, blurring of vision, and metamorphopsia (58).
Ocular examination shows corneal deposits like white-yellow dots seen in 50% of the patients; cataracts are also seen. Fundus changes include decreased foveal reflex initially followed by pigmentary mottling. Classic bull’s eye maculopathy is a sign of late and chronic hydroxychloroquine toxicity, which may lead to permanent pigmentary changes (189).
Diagnostic workup. Macular changes can be detected with the use of autofluorescence, and visual field defects can be assessed with the 10-2 central static threshold test on perimetry. The multifocal ERG (mfERG) may be more suitable for the evaluation of hydroxychloroquine toxicity, but its role as a screening test remains to be established.
According to new screening guidelines, Amsler grid testing or retinal photos lack the sensitivity for the early detection required for effective intervention. Patients should have a 10-2 automated visual field test and one of three objective tests: the macular spectral domain optical coherence tomography (SD OCT), fundus autofluorescence, or mfERG. For a current review of the efficacy of mfERG as a screening test, the reader is referred to Tsang and colleagues who concluded that the mfERG shows a high sensitivity, but variable specificity, when verified against other forms of testing. They further suggested that the greater average cumulative dose in the false-positive group compared with the true-negative group (verified against automated visual field testing) suggests that mfERG may have the ability to detect cases of toxicity earlier than other modalities (212).
Prevention. The daily and cumulative dosage, duration of treatment, coexisting renal or liver disease, patient age, and concomitant retinal disease are risk factors for toxicity (214).
In 2015 a committee constituted by the American Academy of Ophthalmology recommended changes in the screening practices for chloroquine and hydroxychloroquine retinopathy “in light of new information about the prevalence of toxicity, risk factors, fundus distribution, and effectiveness of screening tools” (145). A summary of the major recommendations follows:
Pattern of retinopathy. It is generally understood that the major effect of prolonged use of these drugs is in the perimacular area. A study has shown that among Asian patients, paracentral areas are frequently involved as well, and may be the only affected area (149).
Recommendation. Screening in Asian patients should involve both paracentral and parafoveal areas.
Drug dose. The committee recommends a maximum daily dose of hydroxychloroquine of less than 5.0 mg/kg real weight. The authors point out that there is no similar recommendation for chloroquine, but the older literature suggests using less than 2.3 mg/kg real weight. It these authors’ experience that the most commonly prescribed dose of hydroxychloroquine is 200 mg BID irrespective of weight. The above recommendations would make this a maximum dose for an 80 kg (176 lb) person. Patients with a significantly lower weight should be dosed at a lower rate.
Concomitant tamoxifen use. Concomitant tamoxifen use (for long-term treatment of breast cancer) increased the risk of retinal toxicity approximately 5-fold (150).
Other major risk factors. High dose and long duration of use are the most significant risks. Another major factor is concomitant renal disease.
Screening schedule. According to Marmor and colleagues, “a baseline fundus examination should be performed to rule out preexisting maculopathy. Begin annual screening after 5 years for patients on acceptable doses and without major risk factors” (145). The initial screening must be done after treatment has begun in order to bill for a “high risk medication” examination. These authors recommend annual examinations starting in the first year to ensure that other pathologies do not emerge in the first 5 years.
Screening tests. The recommended primary screening tests are automated, full threshold, visual fields plus spectral-domain optical coherence tomography. These should look beyond the central macula in Asian patients. Modern screening should detect retinopathy before it is visible in the fundus.
Because of the subjective nature of visual field testing and the existence of a significant practice effect, any abnormal visual field should be repeated at least once to ensure reliability. Because of the ubiquitous nature of the Zeiss visual field machine that instrument is recommended by the committee. A 10-2 full threshold test is recommended for evaluation of parafoveal changes. A 30-2 full threshold test is recommended for paracentral changes, particularly in Asian patients. The 10-2 has a radius of 10 degrees around fixation and tests in 2 degrees by 2 degrees grid pattern. The 30-2 has a radius of 30 degrees around fixation and tests in a 6 degrees by 6 degrees grid pattern.
All ocular coherence tomography testing should be done with high definition, spectral domain instruments. The early retinal changes in Plaquenil toxicity are very subtle and every attempt to maintain a high signal strength should be made during testing. This includes dilation and the use of wetting drops in those patients who have “dry eyes.” With the Zeiss ocular coherence tomography instrument, it is these authors’ opinion that a signal strength of 7 or higher is required for the adequate evaluation of the receptor layer.
Additional testing. The multifocal electroretinogram (mfERG) can provide objective corroboration for visual fields, and fundus autofluorescence can show damage topographically. There have been reports that multifocal electroretinography can be very sensitive to early changes in Plaquenil toxicity, but there are concerns about an inherent variability in the ERG data. Fundus autofluorescence has become widely available both in fundus photography and in an ocular coherence tomography format. The autofluorescence derives from the response of lipofuscin in the eye to stimulation. This means that significant retinal changes have occurred at the level of the pigment epithelium before significant fluorescence is present.
Toxicity. According to Marmor and colleagues:
|
Retinopathy is not reversible, and there is no present therapy. Recognition at an early stage (before any RPE loss) is important to prevent central visual loss. However, questionable test results should be repeated or validated with additional procedures to avoid unnecessary cessation of valuable medication (145). |
Tests not recommended for screening. According to Marmor and colleagues (145):
|
• Fundus examination and photography. Ophthalmoscopy is not a screening tool because photoreceptor damage is detectable with other techniques well before visible changes in the fundus. A bull’s-eye, by definition, implies RPE loss and an advanced stage of toxicity. | |
|
• Time-domain optical coherence tomography. The resolution is not sufficient to detect early toxic changes. | |
|
• Fluorescein angiography. This can recognize RPE defects, but these are late changes. | |
|
• Full-field electroretinogram. The full-field electroretinogram is a global test of retinal function and will show abnormalities in only very late chloroquine or hydroxychloroquine toxicity. It may be useful to judge the extent of damage beyond the macula. | |
|
• Amsler grid. Amsler grid testing is not consistent enough for reliable screening of subtle scotomas. | |
|
• Color vision testing. Color errors may occur but are not sensitive or specific. | |
|
• Electrooculogram. The electrooculogram has not been validated as a reliable screening test. |
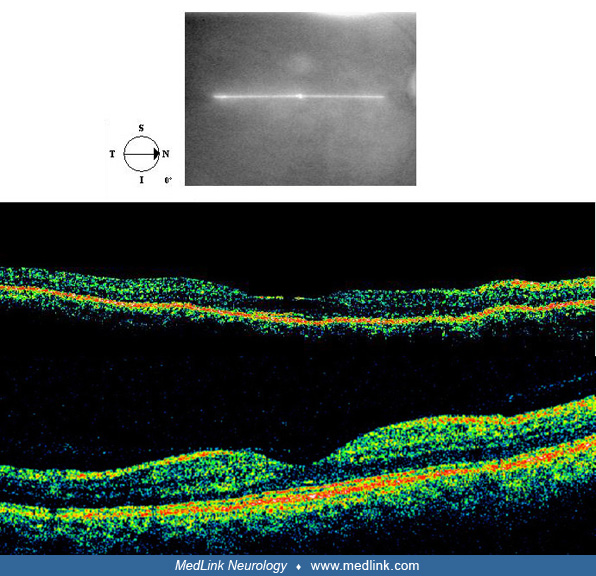
One final note on OCT imaging. The neural retina is approximately 300 µm thick. For reference, that’s the thickness of three sheets of printer paper pressed together. Ocular coherence tomography HD scans can produce cross-sectional views of the retina with almost micrographic detail. (See the normal OCT figure in the Diagnostic workup section above). With this resolution, it is possible to detect preparametric (before visual field loss) damage to the receptor layer of the retina, provided that the scans have sufficient signal strength and the reader is sensitive to subtle disruptions in the two lines just above the retinal pigment epithelium/interdigitization zone. The first of these lines is the ellipsoid zone, which many believed to be the demarcation between the inner and outer segment of the receptors. The very faint line above the ellipsoid zone is the outer limiting membrane. It is these authors’ opinion that any disruption in these two lines represent the earliest changes in the receptor layer and precede total receptor layer loss. The labels given the ocular coherence tomography layers have varied widely as this technology has come online. For a current consensus on ocular coherence tomography HD layer labels the reader is refereed to Staurenghi and colleagues (199).
Prognosis and complications. Prognosis of hydroxychloroquine retinopathy may depend on the severity of the retinopathy when therapy was discontinued. Although pre-maculopathy is mostly reversible and most cases of true retinopathy remain stable, advanced cases may even progress after discontinuation of therapy (143; 144).
Management. Management includes cessation of medication before the retinal pigment epithelium is damaged to a point that leads to significant photoreceptor loss.
Definition. Thioridazine retinopathy has subjective symptoms of decreased visual acuity, problems with dark adaptation, night blindness, and central scotoma (24).
Etiology. Thioridazine is a class of neuroleptic commonly used for treatment of psychiatric disorders. When administered in daily doses of more than 800 mg and for longer than 2 years, it can cause oculogyric crises, miosis, mydriasis, cycloplegia, disturbances of accommodation, and lens and corneal opacities. Symptoms are similar to those in retinitis pigmentosa (62).
Pathogenesis and pathophysiology. The pathogenesis of the toxicity is still unknown. Some hypotheses state that drug absorption leads to choriocapillaris disorganization, resulting in pigment alteration in the retinal epithelium, which alters the nutrition of the photoreceptors. Other theories include changes in the Muller cells and photoreceptors causing phosphorylation due to enzymatic alteration (62).
Clinical presentation. Most commonly the effects of acute toxicity from thioridazine are nyctalopia, blurred vision and dyschromatopsia, and fine salt and pepper pigment retinopathy involving both the posterior pole and the periphery in the fundus with geographic choriocapillaris atrophy (184).
Diagnostic workup. ERG findings are characteristic for diffuse, reduced photopic and scotopic states (184).
Prognosis and complications. Interruption of thioridazine administration can lead to disappearance of the pigmentary changes, regression of the visual subjective symptoms, and normalization of the ERG (62).
Management. There is no specific means of treatment other than discontinuing the drug.
Definition. Necrotizing retinitis is the hallmark of ocular toxoplasmosis caused by the parasite Toxoplasma gondii. It is commonly accompanied by anterior uveitis, vitreous inflammatory reaction, retinal vasculitis, and macular edema (93).
Etiology. Toxoplasmosis is produced by the parasite Toxoplasma gondii, and the cat is the definitive host. Humans and other animals act as intermediate hosts. Congenital toxoplasmosis, as well as toxoplasmosis in immunocompromised patients, is potentially fatal (107).
Pathogenesis and pathophysiology. The definitive host is the cat in which the parasite develops five types of multiplicative and sexual stages in the intestinal epithelium before being eliminated as sporozoites or oocysts in the feces. When infection in humans occurs, it could be by the ingestion of the sporozoite form in improperly cooked meat or congenital by transplacental transmission. The parasite has three forms: tachyzoite (infectious form), bradyzoite, and sporozoite. The infectious form causes multiplication intracellularly and apoptosis of the cell, releasing more tachyzoite. The bradyzoite is found in the tissue cyst caused by the response to the immune system; this cyst can be found in the retina without any clinical manifestation until the cyst ruptures into the surrounding tissues and causes recurrent infection (54).
Millions of sporozoites are released to the environment for the first 2-3 weeks, and this form can remain infectious for up to 2 years.
Histopathological studies have found necrotizing changes with destruction of the retinal and choroidal tissue. Tissue cyst is mainly found in the outer layers of the retina, with infiltrates of lymphocytes, macrophages, and epithelioid cells (215).
Epidemiology. Around 30% of the world’s population is estimated to be infected. Necrotizing toxoplasmic retinochoroiditis has been reported to occur in 1% to 21% of patients with acquired systemic infection (06; 25). It is often asymptomatic in immunocompromised patients, and ocular lesions may be present in up to 20% (93). Visual outcome for blindness is 24%, and this figure seems not to be affected by the use of multiple antiparasite medications. Recurrence rate is 79% after 5 years and 57% within 2 years (25). There is a higher risk for patients with toxoplasmic scars. Mean age is between 15 and 45 years in 65% of the cases. The frequency and incidence of congenital and acute infection is unknown due to the fact that 85% of patients have ocular involvement during the chronic phase, with a rate of 25% for primary ocular lesions; bilateral involvement is seen in 20% to 40%, with presence of macular lesions in 58% of the cases; and some authors report 64% in the periphery. Apparently, recurrence of ocular toxoplasmosis ranges from 40% to 79% in 5 years of follow-up.
An investigation of ocular involvement following an epidemic of postnatally acquired Toxoplasma gondii attributed to a contaminated municipal water supply revealed a substantial risk of ocular involvement with lesions continuing to develop during the first year after infection (190).
Clinical presentation. The clinical manifestations of congenital toxoplasmosis include microphthalmia, nystagmus, and strabismus. Necrotizing retinitis is frequently found, leading to scaring in the macula (31).
The areas of retinitis are usually caused by rupture of the cysts located in the retinal tissue. These lesions measure one or more disc diameter and are surrounded by retinal edema, inflammatory cells, and perivascular exudates peripheral to the area of acute inflammation. Acquired toxoplasmosis patients with necrotizing retinitis present with vitreitis and complaints of pain, blurred vision, floaters, and photophobia (177). Intracranial calcifications may also be present.
Diagnostic workup. Demonstration of Toxoplasma gondii antibodies (IgM) in serologic studies and compatible fundus lesions are needed to confirm the diagnosis of acute infection (17).
Prevention. The risk factors for recurrent ocular toxoplasmosis are not known, and identification of patients who will develop recurrences is at present not feasible (27).
Management. Therapies aim to stop the tachyzoites’ multiplication, but this does not eradicate the parasite. Treatment is usually given for 4 to 8 weeks depending on severity of disease. The most popular regimen, known as ‘‘classic therapy,’’ consists of pyrimethamine, sulfadiazine, and prednisone. ‘‘Quadruple therapy,’’ also a frequently used regimen, consists of pyrimethamine, sulfadiazine, clindamycin, and prednisone. Folic acid antagonists inhibit the DNA synthesis; commonly used is a combination of sulfadiazine 2g for the initial dose, followed by 1 g orally 4 times and pyrimethamine 25 mg 3 times a day for the first day then 25 mg daily, and folic acid 3 to 5 mg twice weekly. Other therapies consist of a combination of sulfadiazine and clindamycin 300 mg orally 4 times a day (197; 119).
A 2013 report by the American Academy of Ophthalmology based on a literature search calls the efficacy of treating ocular toxoplasmosis into question. Eight of the 11 studies reviewed were randomized controlled studies, none of which demonstrated that routine antibiotic or corticosteroid treatment of toxoplasma retinochoroiditis favorably affected visual outcomes. There was some evidence from one study suggesting that long-term treatment with combined trimethoprim and sulfamethoxazole prevented recurrent disease in patients with chronic relapsing toxoplasma retinochoroiditis. Adverse effects of antibiotic treatment were reported in as many as 25% of patients. There was no evidence supporting the efficacy of other nonmedical treatments such as laser photocoagulation (110).
Definition. Secondary syphilis is well known to cause fulminant vitreitis and retinitis. Many patients infected with HIV also have a high incidence of sexually transmitted diseases (123).
Etiology. Treponema pallidum is the spirochete implicated in syphilis. It disseminates widely via hematogenous (able to penetrate intact mucosal membranes) and lymphatic and vascular channels (07).
Pathogenesis and pathophysiology. In pathological studies, T pallidum organisms have been demonstrated to be contained within the phagosomes of epithelial cells, fibroblasts, plasma cells, and the endothelial cells of small capillaries playing an important role in immune invasion and CNS penetration.
Epidemiology. Cases are steadily increasing. Between 2000 and 2004, from 5979 to 7980 cases were reported, an increase of 33.5% from the prior decade (Centers for Disease Control 2004).
Clinical presentation. Primary lesions appear after 21 years of inoculation and may persist for 2 to 6 weeks. Secondary syphilis occurs 6 weeks to 6 months after resolution of the primary lesion. The main ocular lesion seen in syphilis is chorioretinitis, but neuroretinitis has been reported in patients with HIV disease. Forty percent of patients in the second stage of syphilis have cerebrospinal fluid abnormalities suggestive of a breach of the blood-brain barrier. Salt-and-pepper changes in the peripheral retina are associated with congenital syphilis (20). Ocular syphilis is bilateral in 50% of cases. Five percent of the patients show multiple, flat, yellow-white, chorioretinal lesions in the posterior pole and midperiphery; this is mentioned in literature as being specific to ocular syphilitic lesions among HIV-positive or otherwise immunocompromised individuals. Those lesions can be distinguished from acute choroiditis by the small, leopard-spot changes in the retinal pigment epithelium seen on intravenous fluorescein angiography after acute syphilitic posterior placoid chorioretinitis resolution (70). Visual acuity usually is impaired, ranging from 20/30 to light perception, and accompanied by other ocular signs, such as iridocyclitis, vitreitis, papillitis, optic perineuritis, and retrobulbar optic neuritis. Other symptoms may be present, such as malaise, fever, alopecia, maculopapular rash, condylomata lata, and generalized lymphadenopathy (Browning 2000).
Diagnostic workup. All syphilitic retinitis can be associated with neurosyphilis, and the patient should undergo a lumbar puncture with fluorescent treponema antibody absorption testing. CSF findings consistent with neurosyphilis include increased leukocyte counts and positive VDRL or FTA-ABS tests (55).
Differential diagnosis. Cytomegalovirus, toxoplasmosis, herpes zoster retinitis, acute retinal necrosis, and tuberculosis retinitis should all be considered.
Prognosis and complications. The most common complication in ocular syphilis is serous macular detachment. Visual acuity is usually recovered within 2 to 15 months after treatment (53).
Management. Penicillin remains the keystone of the current regimens. In early primary or secondary syphilis, 2.4 million units benzathine penicillin G given once intramuscularly is usually sufficient, with probenecid (500 mg orally 4 times per day) for 10 to 14 days. If the infection is recurrent or if the duration of the infection is greater than 1 year, a total of 7.2 million units should be given (123).
Definition. Cytomegalovirus is the most common infection seen in AIDS patients.
Pathogenesis and pathophysiology. Primary infection is usually unapparent and remains latent within the host, reacting and shedding when the host immune system is compromised. It is contracted from close personal contact and body fluids (92). It is believed that the virus is seeded hematogenously in the retinal tissue by affecting the monocytes, which is facilitated by retinal cells already affected by an infection such as HIV. It spreads over the course of weeks or months throughout the entire retina (52).
Epidemiology. Cytomegalovirus is a prevalent pathogen present in 40% to 100% of the general population showing prior exposure by serology. Some studies state that one third of patients with AIDS will present with cytomegalovirus (56). Before the use of HAART, cytomegalovirus-related retinitis was the most common intraocular infection in patients with AIDS, occurring in up to 40% of patients during their disease.
Clinical presentation. A typical infection consists of acute fever, increased lymphocytes (50%), malaise, chills, sore throat, headache, fatigue, and nonspecific skin rash; elevation of liver transaminase, anemia, thrombocytopenia, and positive cold agglutinins also can be found (183).
Retinopathy caused by cytomegalovirus can be bilateral or unilateral, with multifocal sites of infection in an individual eye. A patient may typically present with painless blurred vision, unilateral floaters, and light flashes. Depending in the location of the lesion, it can present with central scotomas, decreased visual acuity, and metamorphopsia in the case of macular involvement (163).
Clinically there are two presentations of cytomegalovirus:
(1) Classic hemorrhagic type, with the appearance of “crumbled cheese and ketchup” (large areas of hemorrhage with retinal necrosis on the side of each affected vessel).
(2) The granular type in which the lesions are found spread out from a central focus, with a granulose yellow appearance. Also, it has a brush fire appearance given by the thinned retinal behind the advancing borders.
Diagnostic workup. The diagnosis of the cytomegalovirus retinal process is a clinical workup alone. Cytomegalovirus infection can be explored if negative heterophil antibody test excludes Epstein-Barr virus mononucleosis. Serology for cytomegalovirus IgM antibodies can remain increased for up to 1 year. IgG antibodies should continue to increase during acute infection, making this a good method to determine that cytomegalovirus is the cause of fever (116).
Prevention. Daycare workers, especially those who work with children under 2 years of age, are at increased risk of acute cytomegalovirus infection. Health care workers that treat patients with cytomegalovirus infection and family physicians should not be restricted from work if strict hygiene control is applied (15; 23).
Prognosis and complications. Without treatment, vision loss is usually due to optic nerve involvement, macular changes, and retinal detachment (204).
Management. Ganciclovir has been the mainstay of the therapy for symptomatic cytomegalovirus since 1988; with a significant increase on the resistant strains, intravitreal doses are usually maintained at 5mg per Kg every 12 hours for 14 to 21 days, with additional 1g orally 3 times a day (103; 95).
Specific anti-cytomegalovirus therapy can slow the destructive course of HAART (which is a protease inhibitor) and at least two nucleoside analogs (which has led to decreased HIV replication and elevations in CD4+ cell counts) 2 months in patients treated with intravenous ganciclovir or foscarnet, 2 to 4 months with intravenous cidofovir, and up to 7 months with the sustained-release intravitreal ganciclovir implant. Although HAART-associated immune recovery uveitis can occur in eyes with cytomegalovirus retinitis and may diminish vision in some patients, it is used for the prevention of recurrent retinitis (161). For a current review of the status of cytomegalovirus retinopathy and syphilitic retinopathy in the present era of highly active antiretroviral therapy the reader is referred to the work of Butler and Thorne (32).
Definition. Asymptomatic retinal manifestations characterized by cotton-wool spots are seen in AIDS (105).
Pathogenesis and pathophysiology. Increased plasma viscosity and immune-complex deposition are believed to be involved. Studies have shown its relation to decreasing CD4:CD8 ratio (60). The finding of cotton-wool spots is due to swollen axons in the nerve fiber layer of the retina, probably associated with areas of focal ischemic injury (Anonymous 1994).
Epidemiology. More than 900,000 individuals are infected with HIV in the United States, and 40 million individuals are believed to be infected with HIV worldwide. HIV-related retinopathy has been reported in 50% of all patients with AIDS (63).
Clinical presentation. The most common manifestation of HIV-related retinopathy is cotton-wool spots, which are nonprogressive and transient and usually resolves within 6 to 8 weeks (94).
The majority of these patients are asymptomatic; in some reports there have been central scotomas and, with macular involvement, vision loss (102).
Diagnostic workup. The diagnosis is clinical with the help of lab results to confirm HIV diagnosis (120).
Differential diagnosis. Infectious retinopathy and cytomegalovirus should be considered.
Prevention. CD4+ lymphocyte count is usually used to predict the onset of ocular infections in patients with HIV: fewer than 500 cells/mm3 is associated with Kaposi sarcoma, lymphoma, and tuberculosis; fewer than 250 cells/mm3 is associated with retinal or conjunctival microvasculopathy, cytomegalovirus, varicella zoster virus, mycobacterium avium, cryptococcosis, microsporidiosis, HIV encephalopathy, and leukoencephalopathy (28).
Prognosis and complications. These lesions tend to resolve in 6 to 8 weeks. Two of the complications that can present are retinal necrosis syndrome and retinal detachment (44; 50).
Management. No specific treatment is available for HIV-related retinopathy. Studies have discussed positive effects of combined antiretroviral therapy on CD4+ T cell in advanced HIV disease (14).
HIV-associated retinopathies have decreased with the introduction of HAART (75). In a study to determine the prevalence of ocular diseases in patients with human immunodeficiency virus (HIV) in the era of highly active antiretroviral therapy (HAART), the records of 151 patients seen between 2003 and 2011 were evaluated (108). The authors found that the overall prevalence of ocular disease had decreased since the introduction of HAART. However, patients with HIV continue to be predisposed to developing ophthalmic disease at higher rates than the general population. Visual dysfunction remains an important source of morbidity in HIV patients, particularly in those with AIDS.
Definition. According to the Executive Committee of the American Uveitis Society, acute retinal necrosis syndrome is characterized by peripheral necrotizing retinitis, retinal arteritis, and severe inflammation of the vitreous (210).
Etiology. HSV-2 is the most common cause of acute retinal necrosis identified in childhood (128). The incidence of chorioretinitis in neonatal HSV is 4% to 20%.
Epidemiology. Often detected in newborns at 1 or 3 months of age, there are an estimated 500,000 herpes simplex virus cases annually in the United States. HSV inflammatory disease is a leading cause of ocular morbidity in young to middle-aged patients. About 9% of nontraumatic iritis is due to herpes simplex virus. The average age of onset of the virus is 46 years of age (133).
Clinical presentation. Ocular involvement with neonatal herpes simplex virus infection occurs in 17% to 40% of cases. It can manifest as a simple conjunctivitis that usually causes a slowly progressive red eye and blurred vision or as severe necrotizing retinitis, usually bilateral, characterized by a small, punctuate, yellow-white retinal lesion, large exudates, hemorrhages, retinal edema, and perivascular inflammation (128).
HSV-2 is a rare cause of acute retinal necrosis in immunocompetent patients, and it has been reported to occur in the contralateral eye up to 30 years after the initial episode. It is clinically distinct, with a prior history suggestive of reactivation of viral infection or first ocular occurrence; also, it is more frequent at a young age and with a history of herpes at birth, a preexisting chorioretinal scar, and triggering events like trauma or systemic immunodepressive therapies. Herpetic retinopathy is rapidly progressive with loss of vision, retinal detachment, or optic neuropathy. Chorioretinitis may be present with geographic, well-demarcated areas of chorioretinal atrophy with hyperpigmented borders; it is commonly found in the periphery and sometimes can affect the macular area (81).
Diagnostic workup. Polymerase chain reaction is used to confirm the cause of acute retinal necrosis, typically using 200mL of sample.
Differential diagnosis. Cytomegalovirus infection, toxoplasmosis, rubella, and syphilis should be considered.
Prognosis and complications. Visual prognosis is related to the severity of the disease and to the integrity of cortical ocular and neurologic structures.
Management. Treatment of acute retinal necrosis involves prompt initiation of intravenous antiviral medication with acyclovir, ganciclovir, or foscarnet in addition to oral steroids to reduce inflammation (157).
Definition. Severe vitreitis and necrotizing retinitis have been well documented in nonimmunocompromised patients (133; 142).
Etiology. Varicella zoster causes two clinically distinct diseases, one of which is varicella. Varicella is extremely contagious, with a generalized vascular rash. It establishes latency in the neural tissue and can reactivate as the second disease from the dorsal root glia, resulting in herpes zoster, a localized cutaneous eruption accompanied by neuralgic pain that is prevalent in older patients (73). In this virus, the main ocular complication is the varicella zoster virus-associated acute retinal necrosis.
Pathogenesis and pathophysiology. Herpes zoster virus reaches the central nervous system via hematogenous spread, possibly with extension along the nerve pathways in the anterior visual system (221).
Epidemiology. Herpes zoster virus – acute retinal necrosis has been described more frequently in immunocompromised patients. Visual changes are noted weeks after the initial events of herpes zoster, bilateral in 50% of the cases. Initially it involves the peripheral retina, with granular, nonhemorrhagic lesions that rapidly extend to the macula. In patients with HIV the full thickness of the retina is usually involved, resulting in total blindness (160).
|
Acute retinal necrosis |
Progressive outer retinal necrosis |
|
Healthy patient |
Immunodeficient/AIDS patient |
|
Blurred vision, pain, floaters |
Blurred vision, floaters, no pain, peripheral vision loss |
|
Anterior uveitis, prominent vitreitis, patches of retinal necrosis, occlusive vasculitis, scleritis, optic neuropathy |
Minimal inflammation |
|
Better prognosis |
Poor prognosis |
Clinical presentation. The lesions may be unilateral or bilateral with the outer retinal layers, sparing the inner layers until much later in the disease progress (90). The initial lesions may be small yellowish lesions in the outer retina, typically between 100 and 300m (66).
Diagnostic workup. Diagnosis is made clinically, and serologic differentiation can help confirm the diagnosis.
Differential diagnosis. Cytomegalovirus, syphilis, pneumocystis infection, toxoplasmosis, and fungal choroiditis should all be considered.
Management. A combination of ganciclovir or foscarnet together with intravitreal injection of ganciclovir can be used (164).
Autoimmune retinopathies are rare diseases characterized by circulating antiretinal antibodies along with loss of visual function in central or peripheral vision. A fundus evaluation (ophthalmoscopy) can be normal or show only minimal findings. The diagnosis of autoimmune retinopathy is difficult. Currently, the diagnosis is made based on the demonstration of serum antiretinal antibodies and the presence of clinical manifestations (including abnormal ERGs, OCT findings, etc.). The mere presence of these antibodies is not diagnostic. Autoimmune retinopathy can be divided into two groups: paraneoplastic and nonparaneoplastic, with paraneoplastic further subdivided into cancer-associated retinopathy and melanoma-associated retinopathy. Nonparaneoplastic autoimmune retinopathy is more common than paraneoplastic retinopathy (78).
Definition. Systemic lupus erythematosus is a chronic, multisystemic inflammatory disease characterized by hyperactivity of the immune system and prominent autoantibody production. One of the ocular manifestations of this disease is retinal occlusive disease, with a wide variety of complications that can lead to blindness. All of the ocular manifestations are a reflection of the systemic disease.
Pathogenesis and pathophysiology. Histopathologic evidence indicates that autoimmune mechanisms are involved in producing the ocular vascular lesions of lupus and that a true inflammatory vasculitis is present in the pathophysiology of the vasculopathy, which can be multifactorial and involves many antibodies (anticardiolipin and antiphospholipids and others) (201).
Independent of the mechanism, the damage made to the vessel wall results in vascular occlusion and a loss of vascular integrity, which can be manifest in the fundus as hemorrhage or edema, and in the case of choroidal vascular damage or occlusion it produces multifocal serous retinal and retinal pigment epithelium detachments (194).
Epidemiology. The estimated incidence of systemic lupus erythematosus ranges from 1.8 to 20/100,000 per year. Incidence is more frequent in women, with 80% to 90% of all cases having an average onset of age in the mid-30s (12).
Clinical presentation. Ocular manifestations tend to occur in patients who have active systemic disease. Appearance and disappearance of retinal lesions parallel the course of systemic disease (12):
|
• Cotton-wool spots |
The retinopathy of chloroquine or hydroxychloroquine used as treatment in systemic lupus erythematosus causes asymptomatic and usually reversible macular pigment mottling in its early phases and profound irreversible visual loss with a bull’s-eye pattern of pigmentary maculopathy in its later phases (74). The recommendations for screening of chloroquine and hydroxychloroquine retinopathy have been updated by the American Academy of Ophthalmology. The risk of toxicity increases sharply after 5 to 7 years of use or a cumulative dose of 1000 g of hydroxychloroquine. The risk increases further with continued use of the drug. The prior recommendation emphasized dosing by weight. Most patients are now given 400 mg of hydroxychloroquine daily (or 250 mg chloroquine). This dose is considered acceptable, except for individuals of short stature, for whom the dose should be determined on the basis of ideal body weight to avoid overdosage. A baseline examination is advised for patients starting these drugs to serve as a reference and to rule out preexisting maculopathy, which might be a contraindication to their use. Annual screening should begin after 5 years (or sooner if there are unusual risk factors). Screening should include full threshold visual fields designed for macular sensitivity testing (eg, Zeiss Humphrey 10-2.) and either spectral domain optical coherence tomography or fundus autofluorescence testing (144). For a more detailed discussion of this topic, see also “Hydroxychloroquine retinopathy.”
Neuro-ophthalmologic signs and symptoms can also be present as a manifestation in CNS lupus (207):
|
• Ophthalmoplegia |
Both optic neuropathy and retinal occlusive disease can result in optic atrophy. Immune complex vasculitis, lupus anticoagulant, and related antiphospholipid antibodies and possibly antineuronal antibodies play a role in the pathogenesis of these neuro-ophthalmologic complications (46).
Other common ocular manifestations in systemic lupus erythematosus are as follows:
|
• Secondary Sjögren syndrome can produce keratoconjunctivitis sicca that has been associated with the presence of HLA-DRW52 antigen and anti-Ro (SSA) and anti-La (SSB) antibodies (03). | ||
|
• Orbital inflammation can produce episodes of acute painful proptosis and diplopia (148). | ||
|
• Intraocular pressure may be elevated (227). | ||
|
• Discoid lesions of the eyelids can mimic chronic blepharitis. | ||
|
• Episcleritis or scleritis may occur. | ||
|
• Other, less common manifestations may occur, such as: | ||
|
- corneal staining | ||
Diagnostic workup. Ocular manifestation may be the first presentation of systemic lupus erythematosus.
Fluorescein angiography can be of help to demonstrate arterial and capillary nonperfusion, leakage from neovascular fronds, and staining of the walls of involved vessels (129).
Differential diagnosis. Hypertensive and diabetic retinopathy should be considered in the differential diagnosis.
Complications. The potential complications encountered in systemic lupus erythematosus include:
|
• Multifocal serous retinal detachments as result of lupus choroidopathy |
Management and prognosis. The three major classes of drugs used in treating systemic lupus erythematosus are nonsteroidal anti-inflammatory agents, corticosteroids, and nonsteroidal immunosuppressive agents. Combinations of these drugs and other modes of therapeutic intervention have helped increase the rate of survival from 50% after 4 years in 1954 to more than 90% at present. For an up-to-date review of the etiology, epidemiology, clinical features, diagnostic findings, and treatment options for systemic lupus erythematosus, the reader is referred to the review by Gurevitz (83).
This is a group of subacute retinal disorders with normal or minimally abnormal fundus appearance and with moderate or severe visual loss in association with systemic cancer.
When there is no identifiable direct or indirect mechanism, the cancer is considered to be “remotely” affecting the eye, and the resulting syndrome is termed an “ocular paraneoplastic syndrome.” Paraneoplastic syndromes typically present prior to the underlying malignancy. Most patients with paraneoplastic syndromes have antibodies associated with a specific malignancy, which allows the workup to be targeted to specific organs or tissue types. In regard to markers for the two most well-known ocular paraneoplastic syndromes, we have:
|
Anti-recoverin |
Cancer-associated retinopathy |
Small-cell carcinoma |
Cancer-associated retinopathy (CAR). This syndrome is most commonly associated with small-cell carcinoma of the lung.
Symptoms at clinical presentation.
|
• photopsia |
Clinical examination findings.
|
• ring scotomas |
Diagnostic workup.
|
• abnormal cone and rod ERG |
Prognosis and complications. Guarded
Management treatment modalities.
|
• cancer treatment |
Monitoring antibody levels after initiating therapy helps gauge the effectiveness of treatment. Treatment response is variable depending on duration and extent of photoreceptor damage. For a review of the pathogenesis and therapy for CAR, the reader is referred to (187).
Melanoma-associated retinopathy. This syndrome often presents after the diagnosis of melanoma is already established. It is associated mostly with extraocular metastasis. The syndrome is rare and occurs more frequently in males than in females (5:1 ratio).
Symptoms at clinical presentation.
|
• photopsia |
Clinical examination findings.
|
• usually a normal fundus appearance |
Diagnostic workup. Full-field ERG demonstrates markedly reduced b-wave amplitudes. The dark-adapted a-wave amplitudes are normal as in congenital stationary night blindness. Antibipolar cell antibody is present.
Management.
|
• cytoreductive surgery |
Results are variable and often ineffective. Treatment should be directed at reducing overall tumor burden.
For a current review of paraneoplastic retinopathy in the context of autoimmune retinopathy, the reader is referred to (78).
These patients produce antibodies reactive to the optic nerve and retina. The condition is more common in women than in men. Average age of presentation is 50 years. Vision loss is often asymmetric and variable. Ninety percent of patients have ERG abnormalities. Patients generally respond to immunosuppression or immunomodulation. The goal of therapy is to prevent further vision loss. Patients may have any type of antibody--most often anti-recoverin, but they may present with novel antibodies as well. Initial antibody studies may be negative; as the disease progresses studies should be repeated as the antibodies will likely become positive. Sixty-six percent of patients are found to have a systemic autoimmune disease such as:
|
• lupus, rheumatoid arthritis, Sjögren syndrome |
This is a group of chorioretinal inflammatory diseases, mostly of unknown etiology, that have overlapping clinical features.
Etiology. There may be genetic predisposition, with HLA-A-29 seen in 80% to 98% of cases.
Pathophysiology. This syndrome is possibly related to retinal autoimmunity (lymphocyte proliferation to retinal S antigen). Mixed T and B cells are found in choroidal lesions.
Epidemiology. Birdshot chorioretinopathy is uncommon, has late onset (approximately 50 years of age), and is more common in Caucasians and females.
Clinical presentation. Patients present with blurred vision and decreased color vision as well as floaters and night blindness. Clinical examination shows mild iridocyclitis, vitreous cells, and ovoid, cream-colored lesions with indistinct borders in the fundus at the level of the retinal pigment epithelium and choroid.
Diagnostic workup. Fluorescein angiography shows early hypofluorescence and late hyperfluorescence of lesions. ERG shows decreased b-wave amplitude.
Prognosis and complications. Twenty percent of cases may have limited disease; most have multiple exacerbations and remissions. Lesions become atrophic, but do not become pigmented over time. Active disease is complicated by retinal vasculitis, disc edema, or cystoid macular edema. Late in the disease, attenuated vessels, optic atrophy, and neovascularization may be revealed. ERG changes may be the predictor of disease recurrence during treatment tapering.
Management. Initial treatment with systemic steroids may help. If the steroid course lasts more than 3 months, other agents such as cyclosporine with or without mycophenolate or daclizumab may have a steroid-sparing effect. In some cases, IVIG may be a safe alternative.
Etiology. The etiology of these syndromes is unknown.
Pathophysiology. The pathogenesis is unknown. There is possible viral association.
Epidemiology. The average age at onset is 28 years, with a female predominance of 3:1 and a prodromal flu-like illness in 50% of patients.
Clinical presentation. Patients present with acute unilateral blurred vision with central or peripheral scotomas and photopsia. Rarely, there is mild iritis or vitreitis. Fundus lesions are white or light orange spots that are multiple, discrete, and perifoveal in location at the level of the retinal pigment epithelium or deep retina. These lesions are migratory over an interval of days. There may be granular pigment changes in the macula and disc edema (219).
Diagnostic workup. Perimetry shows a big blind spot and scotomas. Fluorescein angiography shows early punctate hyperfluorescence in a wreath-like configuration with late staining of lesions. ERG shows decreased a-wave amplitude, and mfERG shows photoreceptor disturbances.
Prognosis and complications. Most cases have spontaneous visual recovery in 2 to 10 weeks. White spots disappear, but there is persistent retinal pigment epithelium granularity. Rarely the condition is recurrent or bilateral (171).
Management. There is no effective treatment.
Definition. Punctate inner choroidopathy is an uncommon inflammatory multifocal chorioretinopathy that affects mostly young myopic women (224). Punctate inner choroidopathy is considered one of the ‘‘white dot syndromes,’’ which includes such conditions as acute posterior multifocal placoid pigment epitheliopathy, multiple evanescent white dot syndrome, birdshot retinochoroidopathy, multifocal choroiditis with panuveitis, presumed ocular histoplasmosis syndrome, and serpiginous choroidopathy. These conditions are all distinguished ophthalmoscopically by discrete, white-yellow lesions in the inner choroid, concentrated in the posterior pole (224). Punctate inner choroidopathy usually presents as a self-limiting disease with a good visual prognosis; however, severe visual loss can result if choroidal neovascularization occurs following the initial inflammatory process (71).
Etiology. The etiology is unknown.
Epidemiology. Because of its rarity and the fact that many cases are asymptomatic, an accurate estimation of the incidence and prevalence of punctate inner choroidopathy is difficult to make, particularly as only the most severely affected may seek medical care. In a study, 16 cases of punctate inner choroidopathy were diagnosed at the University of Iowa over a 14-year period (29). Assuming a referral base population of approximately 3 million, the national incidence of punctate inner choroidopathy would be 100 to 200 cases per year. In a series of 77 cases of punctate inner choroidopathy, 90% of patients were women and 97% were white, with a median age of 30 years (15 to 55) (71). Most (85%) were myopic, and no clear association was found between alcohol consumption, cigarette smoking, sexual orientation, or recent viral exposure.
Pathogenesis and pathophysiology. Punctate inner choroidopathy has a strong predilection for young females who have a much higher incidence of inherited autoimmune diseases (eg, lupus, rheumatoid arthritis, multiple sclerosis to name a few). It has been suggested that the white dot syndromes may have an autoimmune component. Becker proposed a common genetic hypothesis for autoimmune and inflammatory diseases (19) and theorizes that inflammatory and autoimmune diseases are discrete entities, the presentation of which is based on the interplay of genetics, immune dysregulation, and environmental triggers (104).
Clinical presentation. In the survey of 77 individuals affected by punctate inner choroidopathy, the most common presenting symptoms were scotomas (defects in the visual field), reported by 91% of patients, blurred vision (86%), photophobia (69%), and metamorphopsia (distortions in central vision) (65%) (71). Patients described having symptom-free periods and episodic relapses; 84% of these experienced one to four relapses per year, with 48% lasting for 2 weeks or longer. Only 3% reported a medical history of an autoimmune disease, and 26% reported a family history of autoimmune disease, including rheumatoid arthritis (18%) and systemic lupus (7%) in first- or second-degree relatives. Two participants reported a first-degree relative with a diagnosis of punctate inner choroidopathy. Fundus examination typically reveals multiple (12 to 25), small (100 to 300 um), gray or yellow, opaque round lesions scattered throughout the posterior pole. These lesions are at the level of the retinal pigmented epithelium and usually evolve into atrophic chorioretinal scars that resemble those found in presumed ocular histoplasmosis syndrome (224). There is no iritis, and no vitreous cells are seen.
Diagnostic workup.
Visual field testing. In a study in which 22 of 30 eyes with punctate inner choroidopathy had visual fields done, the most frequent visual field defect detected was enlargement of the blind spot (9 eyes, 41%) (174). Central/paracentral scotomata were detected in 14% (3 eyes). No cecocentral or peripheral scotomata were observed. In many patients, the blind spot extended towards the macula.
Fluorescein angiography. Punctate inner choroidopathy lesions are usually hyperfluorescent in the early arterial phase, with staining observed in the arteriovenous phase. Occasionally, the lesions block fluorescence in the early arterial phase and stain in later stages. More lesions are seen on fluorescein angiography than on clinical examination. As the disease progresses, damage to the retinal pigmented epithelium occurs and fluorescein angiography demonstrates retinal pigmented epithelium window defects (owing to depigmentation of the retinal pigmented epithelium). Leakage of fluorescein into the subretinal space is seen in patients with a serious retinal detachment (224).
Electrophysiology. In an electrophysiological study, 7 out of 16 patients with punctate inner choroidopathy demonstrated normal flash electroretinography (174). Three of the seven patients (42.8%) had mild asymmetry in b-wave amplitudes between the two involved eyes, which correlated with differences in the number of chorioretinal lesions present in each eye.
Differential diagnosis. Multifocal choroiditis with panuveitis, presumed ocular histoplasmosis syndrome, and multiple evanescent white dot syndrome may resemble punctate inner choroidopathy and should be considered in the differential diagnosis of this disease. Other conditions usually included in the differential diagnosis, but which have distinct features, are acute posterior multifocal placoid pigment epitheliopathy, birdshot retinochoroidopathy, and serpiginous choroidopathy.
Prognosis and complications. As stated in the introduction, this disease is usually self-limiting, and no treatment is advised in the inflammatory stage. These patients should be followed with the primary goal of making sure that a choroidal neovascular membrane does not develop. Choroidal neovascular membrane is most likely to occur when the primary lesion is in the posterior pole and close to fixation (eg, macula). Should this be the case, the patient should be instructed in self-testing with an Amsler Grid and encouraged to return for a follow-up examination at the slightest indication of metamorphopsia.
Management. If neovascularization occurs, treatment with anti-vascular endothelial growth factor (VEGF) injections is recommended. Anti-VEGF drugs (bevacizumab) were used in four patients with choroidal neovascular membrane attributable to punctate inner choroidopathy (37). They received monthly 1.25 mg intravitreal bevacizumab injections for 3 months. All patients had visual improvement of at least one line following treatment; 75% had improvement of two or more lines. In another study, 12 eyes of 12 patients with subfoveal or juxtafoveal choroidal neovascularization secondary to punctate inner choroidopathy received intravitreal bevacizumab injections (1.25 mg) (235). After 12 months, all eyes had stable or improved vision, and nine eyes (75%) showed an improvement of two lines or greater. All lesions converted to the cicatricial stage, and no drug-related systemic or ocular side effects were observed.
Photodynamic therapy consisting of an intravenous infusion of the photosensitive drug verteporfin, which is then activated by a laser, results in the occlusion of the treated vessels. Photodynamic therapy has the advantage over conventional laser treatment because of its ability to selectively damage choroidal neovascularization with relatively minor damage to other tissues. The trend has been to combine anti-VEGF drugs with photodynamic therapy in this condition (47; 225).
If there is no choroidal neovascular membrane, the use of steroids (systemic or periocular) may be helpful. The choroidal neovascular membrane needs to be treated accordingly and leads to poor outcome. For a review of punctate inner choroidopathy, the reader is referred to (08).
Etiology. The etiology is unknown, although possibly an infectious agent (viral) is implicated. An autoimmune etiology is also suspected.
Pathophysiology. There are two theories regarding pathophysiology. The retinal pigment epithelium theory maintains that primary damage is at the level of the retinal pigment epithelium layer and, hence, is primarily retinal in origin. The choriocapillaris theory states that the primary damage is in the choriocapillaris layer; hence, the damage is primarily choroidal in origin. It is associated with HLA-B7 in 40% and DR2 in 57% of cases.
Epidemiology. APMPPE typically presents in young adults, with equal male-female incidence and more commonly in Caucasians.
Clinical presentation. The disorder has two manifestations:
• Systemic. Systemic presentation includes headache and nonspecific viral-like prodrome (50%). There is also cerebral vasculitis with meningeal signs and cerebrospinal fluid pleocytosis. | |
• Ocular. The ocular form presents with acute bilateral symmetric loss of vision, which is temporary and accompanied by the presence of multiple large, yellow-white plaque-like lesions at the level of the retinal pigment epithelium or choriocapillaris in the posterior pole. |
A publication distinguished four phases of APMPEE based on multimodal imaging: choroidal, chorioretinal, transitional, and resolution. Lesions in the choroidal phase can resolve without any morphological sequelae. In the chorioretinal phase, there is evidence of active lesion on fluorescein angiography with early hypofluorescence and late hyperfluorescence, persistent hypocyanescence of lesions across indocyanine green angiography frames, and choroidal hypoperfusion on optical coherence tomography angiography.
Diagnostic workup. Characteristic fundus lesions on fluorescein angiography classically block early and stain late. Indocyanine green angiography shows marked choroidal hypofluorescence in the acute stages. ERG amplitudes may be decreased during the acute stage. Optical coherence tomography angiography enhances understanding of the hypoperfusion choriocapillaris involvement of lesions, noninvasively.
Prognosis and complications. Acute lesions resolve within several days to 5 weeks, with residual retinal pigment epithelium changes. Lesions may become confluent. Visual prognosis is good as 80% to 90% of patients recover to visual acuity of 20/40 or better (228). Visual field defects may remain, however. Neurologic signs and symptoms, especially headaches, are frequent in APMPPE and should be taken seriously. Adequate investigations, including MRI and CSF examination, should be initiated in these patients (208).
Management. In most cases, no treatment is needed.
Etiology. The etiology is unknown.
Pathophysiology. HLA-B7 is present in 55% of cases. There is diffuse lymphocytic infiltration of the choroid and the retinal venules with atrophy of the retinal pigment epithelium.
Epidemiology. The disease affects middle-aged adults (average age, 48 years), with equal male to female incidence. All races are affected.
Clinical presentation. The disorder affects healthy adults that present with progressive and recurrent, bilateral, permanent loss of vision. Fundus exam shows large, peripapillary, pseudopodal, yellow-gray lesions at the level of the retinal pigment epithelium and choriocapillaris when active. Inactive areas appear as well-defined snake-like areas of atrophy and hyperpigmentation of the retinal pigment epithelium (134).
Diagnostic workup. Fluorescein angiogram shows active lesions blocking early and staining late. ERG amplitudes are diminished.
Prognosis and complications. The disease is progressive and medically resistant. It results in permanent scotomas. Prognosis is worse if there is associated choroidal neovascularization, which occurs frequently. Visual acuity is reduced to 20/200 in both eyes for 5% of patients and in one eye for 40% of patients.
Management. Most treatments are ineffective. Triple agent therapy (cyclosporine A + azathioprine + prednisone) has been used with questionable success.
Definition. Acute zonal occult outer retinopathy (AZOOR) is a rare and usually bilateral disease of unknown etiology characterized by focal degeneration of photoreceptors (156). A total of 131 cases of AZOOR (205 eyes), including the variant known as acute annular outer retinopathy, have been reported in the English-language literature. The chief complaint is usually that of an acute-onset scotoma and photopsia. Visual acuity is often normal or nearly so. The fundus most frequently appears normal. Blind spot enlargement in the visual field is a frequent occurrence. Typically, patients retain good visual acuity (20/40 or better). It is unusual for visual field loss to continue beyond 6 months. Early symptoms include photopsia with movement of colors or lights and acute visual field loss in one or both eyes, usually near the blind spot or in the temporal visual field. The relative afferent pupillary defect is minimal or absent despite large scotomas that are asymmetric between eyes (67). Although an association between AZOOR and central nervous system demyelinating lesions has been suggested, misdiagnosis of AZOOR as optic neuritis in not uncommon.
Etiology. The etiology of AZOOR is unknown. Gass speculates that AZOOR belongs to a spectrum of diseases that includes multiple evanescent white dot syndrome, punctate inner choroidopathy, and multifocal choroiditis and panuveitis, among others (69).
Pathogenesis. The pathogenesis of AZOOR is not known. Two hypotheses have been put forward. Gass believes it to be a primary infection of retinal photoreceptors, probably viral, with a secondary host immune response mounted against the infected cells (68). Jampol and Becker argue for an autoimmune/inflammatory cause (104).
Epidemiology. AZOOR occurs in women over men 3:1 and in whites over other races 9:1. Most patients with AZOOR are young adults, with a mean age of 37 years (156).
Clinical presentation. Gass considers the defining characteristic of AZOOR to be acute onset of decreased vision in a zone of the visual field, often accompanied by photopsia (68). At the time of initial diagnosis, the fundus presentation is frequently normal (90% of patients), though retinal pigment epithelial disturbances often appear with time. Electroretinographic abnormalities were found in 99% of patients tested. The multifocal ERG is abnormal in the retinal zone corresponding to the area of visual field loss, confirming that the abnormality is retinal and not the result of an optic neuropathy. Of the patients followed by Gass, 20% reported a viral-like illness shortly before the onset of symptoms. A third of patients have an associated autoimmune disease, such as Hashimoto thyroiditis or relapsing transverse myelopathy. Gass and colleagues found that the median delay in the diagnosis of AZOOR was 17 months.
Diagnostic workup. Visual field and electroretinographic testing are the two tests that are essential to the diagnosis of AZOOR. Fundus fluorescein angiography appears to have limited usefulness. Indocyanine green autofluorescence fundus photography and indocyanine green angiography have been done in small numbers of patients, also with limited results. High-resolution optical coherence tomography shows areas of defects in the boundary between the inner segments (IS) and the outer segments (OS) of the photoreceptors, termed the “IS/OS boundary,” which clinically corresponds to blind spot enlargement seen in visual field testing. This characteristic finding appears to be the most promising of the newer diagnostic procedures (61).
Differential diagnosis. The differential diagnosis includes autoimmune retinopathy, cancer-associated retinopathy, and melanoma-associated retinopathy (89), as well as all of the white dot syndromes, a group of disorders characterized by multiple whitish-yellow inflammatory lesions located at the level of the outer retina, retinal pigmented epithelium, and choroid. For further explanation of these differential diagnoses, the reader is referred to an excellent review of AZOOR by Monson and Smith (156). Relative afferent pupillary defect (RAPD) is a principal parameter for differentiating optic neuropathies from retinal disorders, and it has been identified in 96% of patients with optic neuritis. However, relative afferent pupillary defect was recorded in 24% and 42.9% of patients with AZOOR in series by Gass and by Jiang and colleagues, respectively.
Management. No treatment has been proven to improve the visual outcome of AZOOR. Systemic corticosteroids are the therapy that has been most commonly attempted, and one reported case supports this approach (113). Fortunately, AZOOR is often self-limiting, and the patients are frequently left with reasonably good visual function. Stabilization of visual field abnormality and central vision loss occurs in 80% of patients within the first 6 months.
Clinical vignette. A 29-year-old Asian female complained of sudden onset of a central scotoma in her left eye; she’d first noticed the central scotoma four days prior to visiting her eye specialist. The scotoma was stable for 3 weeks. The patient also complained of intermittently flashing lights, which eventually became constant, in the left temporal visual field.
|
Ophthalmic exam: | ||
|
Best corrected central visual acuity: |
Right eye: 20/20-3 | |
|
Left eye: count fingers at 1 ft | ||
|
Color vision: |
Right eye: normal | |
|
Left eye: unable to identify Ishihara plates | ||
|
EOM: |
No signs of CN paresis, internuclear ophthalmoplegia, or pathological nystagmus | |
|
LIDS: |
Normal | |
|
PUPILS |
Round, (+) 0.9 log unit relative afferent pupillary defect left eye | |
|
Anterior segment exam: |
Normal Intraocular pressure: TA: 17 mmHg both eyes (normal below 22). | |
|
Fundus exam (dilated): |
Optic disc: healthy rims with cup-to-disc ratio 0.35, both eyes. | |
|
Macula: appears normal with no edema or exudates, both eyes. | ||
|
Periphery: normal, both eyes. | ||
|
Visual field testing (Humphrey automated perimetry) |
Right eye: normal visual field | |
|
Left eye: dense scotoma inferotemporal | ||
|
Full field ERG |
Normal | |
|
VEP: |
Right eye: normal | |
|
Left eye: extinguished | ||
Initial diagnosis was optic neuritis. MRI of brain was normal. The patient’s vision worsened, so the patient sought the opinion of a neuro-ophthalmologist who performed a multifocal ERG. The mfERG showed normal responses in the right eye and depressed responses in the affected left eye. An indocyanine green angiography was subsequently performed and showed normal findings in the right eye. Abnormal indocyanine green angiography in the left affected eye revealed pigmentary choroidal disturbances. Final diagnosis was acute zonal outer retinopathy.
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.
Rosa Ana Tang MD MPH MBA
Dr. Tang of Neuro-ophthalmology of Texas, PLLC, has no relevant financial relationships to disclose.
See ProfileJade S Schiffman MD
Dr. Schiffman, Director of the Eye Wellness Center in Houston, Texas, has no relevant financial relationships to disclose.
See ProfileFrancisco R Sanchez Moreno MD
Dr. Sanchez Moreno of Mayo Clinic School of Graduate Medical Education has no relevant financial relationships to disclose.
See ProfileRicaurte Crespo MD
Dr. Crespo of Neuro-Ophthalmology of Texas in Houston has no relevant financial relationships to disclose.
See ProfileJames W Walters PhD OD
Dr. Walters of the University of Houston has no relevant financial relationships to disclose.
See Profile
Heather E Moss MD PhD
Dr. Moss of Stanford University has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
General Neurology
Jan. 13, 2025
General Neurology
Jan. 13, 2025
Neuro-Ophthalmology & Neuro-Otology
Jan. 08, 2025
Neuro-Ophthalmology & Neuro-Otology
Jan. 07, 2025
General Neurology
Dec. 30, 2024
Neuro-Oncology
Dec. 13, 2024
General Neurology
Dec. 13, 2024
General Neurology
Dec. 13, 2024