








Neuro-Oncology
Choroid plexus tumors of childhood
Jan. 14, 2025
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Worddefinition
At vero eos et accusamus et iusto odio dignissimos ducimus qui blanditiis praesentium voluptatum deleniti atque corrupti quos dolores et quas.
X-linked myotubular myopathy is a form of centronuclear myopathy mainly affecting newborn boys, but it can sometimes also affect girls and adults. Diagnostic methods have improved so that the majority of patients and carriers may now have their diagnosis verified by molecular methods. Currently, the complex pathogenetic mechanisms are better understood and include defects in membrane tubulation and excitation-contraction coupling, providing evidence towards a common basis for the X-linked and autosomal forms of centronuclear myopathy. Several studies of potential therapeutic modalities aimed at ameliorating disease severity have reached proof of concept, natural history studies have been completed, a number of animal models have been established and experimental trials carried out, outcome measures have been defined, and therapeutic trials are being planned. These trials have investigated inventive methods, such as therapies based on antisense oligonucleotides, gene therapy, and drug repurposing. Although the first in-human gene transfer study was affected by a major drawback, these innovative initiatives are leading the way towards a future of hope.
|
• X-linked myotubular myopathy is caused by mutations in the myotubularin gene MTM1. | |
|
• Molecular genetic verification is often possible through sequencing, but it should be noted that copy number variations may also be causative, as may intronic alterations, and this may require other analytic methods. | |
|
• Female carriers may present with overt disease. | |
|
• Although no curative treatment exists to date for this usually very severe disorder, active treatment is indicated, at least initially, because of the favorable course in some neonatally severe cases. | |
|
• Hepatobiliary disease is quite common and should be carefully monitored. | |
|
• Prognosis cannot be based solely on the nature of the mutation, and decisions about treatment have to be taken on an individual basis. | |
|
• Differential diagnoses include the autosomal forms of centronuclear myopathy and myotonic dystrophy. | |
|
• Most, but not all, mothers of affected boys are mutation carriers; mosaicism has been reported. |
X-linked myotubular or centronuclear myopathy is assigned to the group of congenital myopathies (OMIM #310400) (69). These myopathies were defined after the advent of histochemical staining methods of muscle biopsy sections on the basis of structural abnormalities in the muscle fibers.
Centronuclear myopathies are characterized by the presence of centrally located nuclei in muscle fibers. This term is used to describe a diverse collection of individually rare congenital myopathies. These conditions are primarily caused by pathogenic variants in MTM1 (approximately 50%), DNM2 (approximately 15%), or BIN1 (approximately 3%). Although a significant percentage (approximately 20%) of centronuclear myopathy cases have an unknown genetic basis, another portion (approximately 12%) involves other genes (24). The centronuclear myopathy–like phenotype has been reported to be associated with pathogenic variants in genes, such as ryanodine receptor 1 (RYR1) (238), titin (TTN) (42), SPEG (01), ZAK (225), DHPR (CACNA1S) (191), and potentially CCDC78 (151) and myotubularin-related protein 14 (MTMR14) (217). Centronuclear myopathies, despite their diverse presentations and modes of inheritance, often show noticeable weakness in the eye muscles, such as ptosis and ophthalmoparesis.
X-linked myotubular myopathy typically represents the most severe manifestation within the spectrum of centronuclear myopathies (39).
In 1966, findings of muscle pathology were reported in a 12-year-old boy who exhibited generalized muscle wasting and facial weakness, including ptosis (202). The authors of this report were surprised to find that the images of the affected muscle fibers closely resembled fetal myotubes. These myotubes appeared slightly elongated, having a central nucleus with one or a few myofibrils. The authors hypothesized that this condition arose from an interruption in the development of muscle fibers and proposed the term “myotubular myopathy” to describe the condition. Although the exact nature of this disease is not yet fully understood, "myotubular myopathy" remains the designated term for the X-linked inherited type of centronuclear myopathy.
In 1969, the X-linked form of myotubular myopathy was described in a large Dutch pedigree in which the affected males showed muscle fibers resembling fetal myotubes (224). A long-term follow-up study of this and another Dutch family shows wide variability in the clinical picture (12), confirmed by reports of patients diagnosed as adults (109; 169).
The coining of the alternative name "centronuclear" myopathy reflects the argument about whether the basic defect in this disorder is an arrest of maturation of the fetal muscle fibers or whether the central nuclei constitute the main histologic feature.
In 1985, eight patients with muscle biopsies showing a centronuclear myopathy pattern were reported (102). The authors of this report proposed three inheritance patterns in this group of patients. They observed that the individuals they identified as having X-linked inheritance were male and displayed a severe phenotype, including neonatal asphyxia, difficulties with breathing and feeding shortly after birth, as well as weakness in the face and limbs during the neonatal period, often leading to early mortality.
In 1996, a consortium of three groups found mutations within the MTM1 gene located on the X chromosome as the underlying cause of this condition (135). The final product of this gene, known as myotubularin (MTM1), is recognized as an enzyme found in endosomes. It plays a role as endosomal lipid phosphatase, which regulates membrane trafficking by modulating phosphoinositide (PI) dephosphorylation and contributes to the structure and function of muscle fibers.
The first disease model of X-linked myotubular myopathy using knockout mice was established in 2002 (38), followed by the development of other animal models (67; 170) and natural disease models in canines (15; 87; 165). These initiatives have led to a better understanding of disease pathogenesis and treatment targets.
Autosomal forms with similar histology have been described (OMIM #60150, #255200). They usually present later and follow a milder course than the X-linked form (232; 112), in dogs also (199), but exceptionally severe cases similar to the X-linked form have also been reported (119; 26). Mutations in the dynamin 2 gene (29) are a common cause of the dominantly inherited form (76; 28; 72; 192), whereas mutations in the ryanodine receptor gene RYR1 have also been identified, including recessively inherited mutations (120; 238; 23; 119).
The first causative gene for autosomal recessive centronuclear myopathy was identified as amphiphysin 2 (BIN1) (162). Subsequently, a patient with internalized nuclei, the histopathology resembling centronuclear myopathy, was found to have compound heterozygous mutations in the titin gene TTN (42; 64). A further differential diagnosis may be myopathy caused by SPEG mutations, often accompanied by cardiomyopathy (01; 235; 236).
It is to be noted that female carriers of the X-linked form can manifest muscle disease, as index patients, also, with no affected males known in the family (100; 230; 205; 115; 128; 190; 94; 169; 24; 103; 119; 74; 189; 27; 83; 242; 48; 126). A female patient described with cardiomyopathy requiring heart transplantation had not undergone analysis of the myotubularin gene.
Onset is perinatal. Polyhydramnios and premature birth are common features. The affected, most often male, newborn presents with severe generalized muscle hypotonia and weakness associated with respiratory insufficiency (13). Many of these boys lack spontaneous antigravity movements at birth, and many fail to establish spontaneous respiration. Feeding difficulties and ophthalmoplegia are common. The affected boys are often long for their gestational age and have relatively low birth weights; large heads, an uncommon characteristic among other congenital myopathies; and long fingers. Tendon reflexes are mostly absent. There may be contractures of the hips or knees. Some boys have cryptorchidism.
Patients diagnosed with X-linked myotubular myopathy are categorized into mild (no ventilation support), intermediate (ventilation support < 12 hours/day), or severe (ventilation support ≥12 hours/day) phenotypes depending on the extent of ventilator assistance needed (155).
Insights from natural history studies unveiled that most patients encounter the severe form of this condition, often displaying profound hypotonia, significant respiratory insufficiency, and difficulties in swallowing at birth.
Most patients exhibit a low Apgar score within the first minute after birth. One of the main initial signs of X-linked myotubular myopathy is the inability to establish spontaneous breathing, resulting in an immediate requirement for respiratory support of patients from birth onwards (16; 155). Some individuals may achieve independent breathing shortly after birth, yet some might lose this capacity around the median age of 6 months, particularly in the severe phenotype group (09). Achieving motor milestones is typically delayed across different phenotypes, and achieved milestones are often lost in patients with the severe phenotype (09).
There is usually no primary cardiac involvement, but a report describes complete atrioventricular block in the absence of cardiomyopathy in a patient confirmed by mutation analysis to have the X-linked form of myotubular myopathy (107). A report described cardiac aneurysms in a patient with an MTM1 mutation; the causal relationship between the mutation and the cardiac disorder, however, was uncertain (20). An exceptional patient showed progressive dementia (154), and a twin pair presented with hypoxic encephalopathy (44).
A few affected boys have been described as showing abnormalities in neuromuscular transmission, and some have responded to pyridostigmine treatment (176).
Usually, but not invariably, the disease follows a fatal course. Formal clinical and histologic criteria for the diagnosis have been defined by the International Consortium on Myotubular Myopathy (233). Clinical reviews are available (232; 117; 140), and McEntagart and colleagues addressed genotype-phenotype correlations (155). Clinical papers describe Danish, German, and Dutch series (237; 82; 175).
There are less severe forms of X-linked myotubular myopathy, with symptom onset ranging from birth to later in childhood (09; 25). Reports highlight varying combinations of signs and symptoms in these patients, encompassing neonatal hypotonia, feeding difficulties that may improve in time, delayed motor milestones, mild to moderate weakness, gait abnormalities, speech difficulties characterized by a weak voice, and recurrent respiratory tract infections (25; 11). In about 20% of all X-linked myotubular myopathy cases with milder presentations, ventilatory support may not be necessary during childhood. However, some patients may experience nocturnal hypoventilation and may require BiPAP (25; 06; 09).
In some of the patients surviving long-term despite neonatally severe disease, involvement of organs other than muscle may ensue. Reports of such manifestations have included pyloric stenosis, spherocytosis, gallstones, hepatic peliosis, intrahepatic cholestasis, kidney stones, vitamin K–responsive bleeding diathesis, nontraumatic subdural or intracranial hemorrhage, and recurrent pneumothorax (105; 125; 144; 161; 158; 239). Among these, hepatic peliosis and other hepatobiliary diseases have been repeatedly reported (98; 159; 81; 55; 158). In one case, peliosis was successfully treated using hepatic artery embolization (213) and in another by liver transplantation (167). There are, however, a number of reports of patients in whom no long-term complications have been noted (237; 06; 09).
Many female cases have been described, with or without evidence of skewed X inactivation (232; 208; 100; 230; 205; 115; 128; 190; 94; 74; 189) and may be late (169; 103). Female patients, who may be regarded as being mosaic in that they have one normal MTM1 allele and one carrying the mutation, mutually exclusively active in any single cell, may show asymmetry and local involvement without showing detectable skewing of their X-inactivation. This is evident not only in terms of muscle weakness but also in terms of growth of the left and right side of the body, including the face and limbs (27; 242; 48). Quite often, the manifestations are milder than in affected males carrying the same mutation (27; 174). Killer cell immunoglobulin-like receptor variants were suggested as possible genetic modifiers of manifestation in female carriers (201). Cocanougher and coworkers developed a clinical classification for manifesting female carriers (48).
The prognosis is usually poor, and most affected boys die in infancy. A number of patients with severe neonatal floppiness and serious respiratory problems have, however, survived. Some of these have even done well, without any residual respiratory difficulty or severe disability (155).
Historically, it was estimated that nearly half of infants did not survive beyond 18 months (155; 16; 92). However, subsequent reports suggest a lower mortality rate, around 10% per year, emphasizing the relative stability in the disease course and the pivotal role of intensive medical intervention in determining survival. The majority of infants necessitate prolonged stays in the neonatal intensive care unit, with about 30% to 50% of their first year spent within hospital. About a quarter of male infants do not survive beyond their first year of life (65).
A considerable number of individuals who survive X-linked myotubular myopathy often contend with disabilities, exhibiting a significant reliance on technology. Nearly 80% of these individuals depend on forms of technology, including 24-hour ventilatory support via tracheostomy, gastrostomy feeding tube, and wheelchair support (06).
The duration of ventilator support is a reliable, consistent, and clinically significant measurement for assessing disability among patients with X-linked myotubular myopathy (06; 66).
The persistent risk of premature death, primarily due to respiratory complications, remains notably high at about 10% annually after infancy. Episodes of hypoxia can arise, potentially resulting in hypoxic-ischemic encephalopathy (65).
Patients with X-linked myotubular myopathy experience frequent hospitalizations, often linked to respiratory failure, respiratory distress, infections (particularly lower respiratory tract infections), as well as various surgical and medical procedures (16; 91).
Patients often undergo various surgeries, including esophagogastric fundoplasty, tracheostomy, myringotomy (with or without ear tube insertion), bronchoscopy, laryngoscopy, orchidopexy, spinal fusion, adenotonsillectomy, and laparoscopy. Gastrointestinal tube insertion is reported to be more prevalent among patients under 18 months old. Procedures, such as gastrostomy, circumcision, muscle biopsy, myringotomy, inguinal hernia repair, and ventriculoperitoneal shunt, are reported at similar rates across both age groups (16).
Scoliosis commonly emerges during later childhood, affecting around 75% of individuals with X-linked myotubular myopathy (65). Long-term survivors have been reported to experience complications, such as pyloric stenosis and cavernous hemangiomas of the liver (105).
Hepatobiliary problems, particularly cholestasis, are common comorbidities among individuals with X-linked myotubular myopathy and require close observation (89). These issues might appear early, particularly after infections or vaccinations, and manifest as elevated liver enzymes and irregularities detected in liver ultrasound, such as hepatic peliosis or gallstones. In the INCEPTUS natural history study, 91% of 34 patients diagnosed with X-linked myotubular myopathy either had a history of liver disease at enrollment (eg, hyperbilirubinemia and intermittent or persistent cholestasis) or exhibited at least one sign of hepatic disease throughout the study period, ranging from 0.5 to 32.9 months (66). Half of the patients required treatment for cholestatic or other liver conditions, and one patient died due to hepatic peliosis. Among the complications associated with X-linked myotubular myopathy, hepatic peliosis emerges as one of the most serious issues not directly related to muscle involvement (16; 158). It is characterized by multiple sinusoidal blood-filled cysts within the liver (159). This complication may affect up to 5% to 10% of individuals (105; 16). Liver hemorrhage or bleeding to the peritoneal cavity caused by this vascular lesion could potentially lead to fatal outcomes.
Rare patients have shown mild disease since the neonatal period (25; 43; 240), and some patients have been diagnosed only as adults (109). Care is to be taken in prognosticating based on the mutation alone (52).
A follow-up study and a few case reports suggest that long-term survivors may be at risk for medical complications involving organ systems other than muscle tissue (see above) (105; 144; 125; 158; 239), but a number of adult patients have been described not showing any of these (12; 25; 240). A mouse model with a lifespan of more than 50 weeks also does not show extra-muscular manifestations (170).
The cause of death is usually respiratory failure, sometimes in combination with pneumonia or cardiac failure.
This fictitious but representative patient was born as the third child of a couple with a history of two male miscarriages. The pregnancy was complicated by polyhydramnios. The male, floppy infant was delivered at term. Birthweight was 3000 g, length was 53 cm, and head circumference was 37 cm. The neonate failed to establish spontaneous respiration, and antigravity movements were few and feeble. An ultrasound scan of the child’s head failed to reveal a central cause for the floppiness. An EMG showed spontaneous fibrillations, and a clinical diagnosis of spinal muscular atrophy was made. A muscle biopsy, however, showed changes compatible with myotubular myopathy, and genetic testing for spinal muscular atrophy was negative. Myotonic dystrophy was excluded by molecular genetic testing. The diagnosis of myotubular myopathy was made. A mutation identified in the MTM1 gene confirmed that the disorder in this family was X-linked. The mother, grandmother, and an aunt were later found to be carriers of the disease-causing mutation. The infant was mechanically ventilated and showed temporary improvement by establishing antigravity movements of the limbs. Doctors weaned him off the ventilator for 2 weeks, but he died at the age of 2 months of pneumonia and respiratory insufficiency. Prenatal diagnosis based on mutation detection could be offered in any future pregnancy of this couple.
Biological basis. This disorder follows X-linked inheritance, with mutations found in the MTM1 gene encoding a lipid phosphatase, myotubularin (MTM1) (135; 59; 134; 63; 163; 207; 36; 60; 131). Hitherto, no other X-chromosomal gene has been implicated.
Note: Autosomal forms with similar histology, and female patients with MTM1 mutation, have been described, and the X-linked mode of inheritance needs to be confirmed by mutation detection. The first gene for an autosomal form was published in 2005 (29) and was followed by three more (120; 162; 42).
Our understanding of X-linked myotubular myopathy is increasing based on the identification of the disease-causing gene, mutation detection in a great number of patients and observed genotype-phenotype correlations. At the same time, basic scientific advances and animal models are clarifying the pathogenetic mechanisms for both the X-linked and the autosomal forms of centronuclear myopathy characterized to date, with a number of molecular links being identified between these disorders (04; 67; 118; 219; 07; 138; 08; 75; 114; 87; 10; 89).
A number of patients with or without MTM1 mutations have been described as showing electrophysiological findings suggestive of a neuromuscular transmission defect, leading to a clinical suspicion of myasthenia (147; 176). Some of these patients showed a response to treatment with acetylcholinesterase.
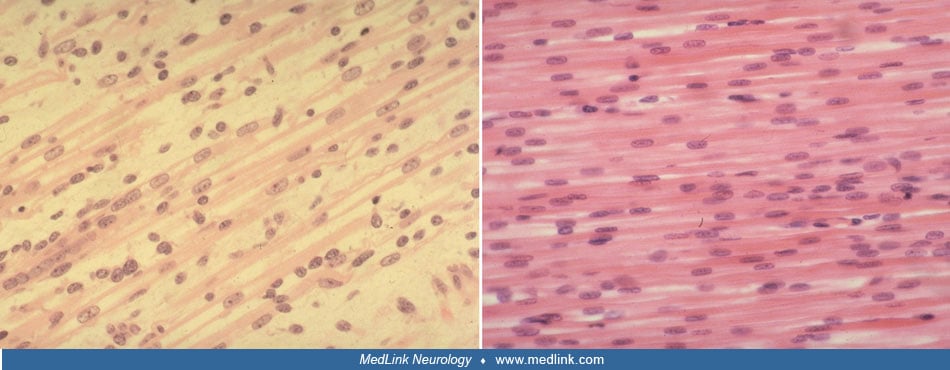
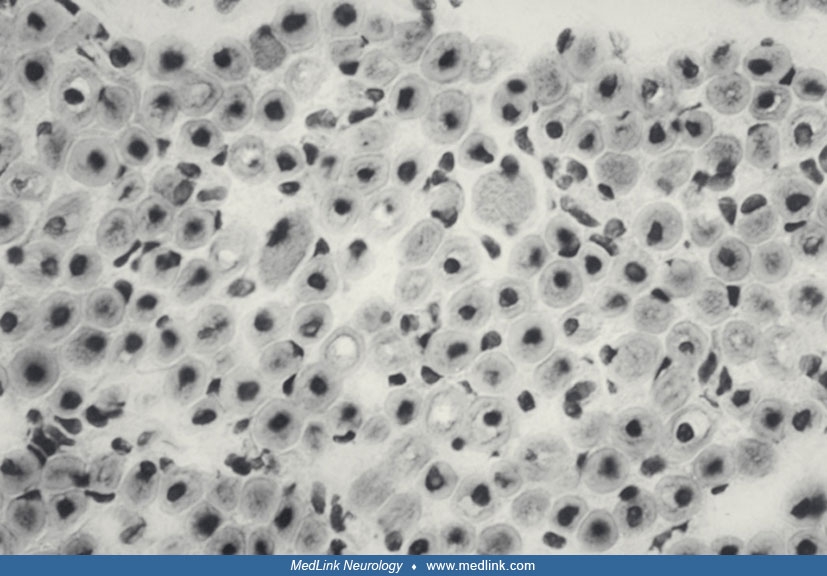
Pathology. Histologic criteria for X-linked myotubular myopathy include the presence of numerous small muscle fibers, many of which contain rows of regularly spaced central nuclei resembling normal fetal myotubes (118). A study addressed the relationship between nuclear mislocalization and fiber contractility in X-linked myotubular myopathy (180). Type 1 slowly contracting fiber predominance is common, and type 1 fiber hypotrophy is observed in some, but not all, cases (Imoto and Nonaka 2001; 118). The myotube-like fibers show a central aggregation of mitochondria, resulting in a highly dense oxidative enzyme staining centrally. The ATPase reaction demonstrates a corresponding lack of central staining. Protein- and cDNA-based methods have identified absence or low levels of myotubularin expression in most patients tested (132; 137; 178; 218). A study described cellular, biochemical, and molecular changes as likely consequences of epigenetic modifications in muscles from patients with X-linked myotubular myopathy (10).
The presence of necklace fibers has been observed in certain cases of late-onset X-linked myotubular myopathy (22) and female carriers (27). These patients initially show mild symptoms during childhood but experience a worsening of symptoms during the first and second decades of life. Necklace fibers, observed through periodic acid–Schiff, oxidative staining, and electron microscopy, are basophilic rings beneath the sarcolemma that follow the cell's contour and align with internal nuclei.
Genetics. The gene for X-linked myotubular myopathy was assigned in 1987 to the proximal Xq28 region (214; 57; 145; 203; 215; 146; 111). The determination of the breakpoints of a deletion in a female patient with X-linked myotubular myopathy (54) and the physical mapping of deletions in two male patients with myotubular myopathy and ambiguous external genitalia helped to narrow down the region likely to contain the disease gene (110). These two male patients were later found to have unusually large deletions, including an alternatively spliced gene in proximal Xq28 (136). Bartsch and colleagues published a description of a familial deletion associated with male hypogenitalism (14). A further patient with a well-characterized deletion in this region had hypospadias and scrotal hypoplasia (189). In contrast, another patient described with a big deletion had normal genitalia (221).
This led to the characterization of a gene in this region (MTM1) showing mutations in affected males (135; 59; 134; 63; 163; 207; 36; 60; 209; 131; 24; 05). The gene encodes a lipid phosphatase, myotubularin. The MTM1 gene has 15 exons, and various disease-specific alterations have been found in most of them (209; 131; 106; 152; 221). The first exon of the MTM1 gene does not consist of coding sequences and serves as a putative promoter region. The start codon is located in exon 2. The complete genomic structure of the human myotubularin gene has been determined (133), and the corresponding sequences are available in GenBank under the accession number AH006018.2.
The incidence of new mutations appears not to be as high as previously thought, and a majority of mothers of affected boys will be found on genetic testing to be carriers (134; 131; 106; 21; 24; 119; 149; 189). It has also become evident that many female carriers manifest symptoms (174) and may present as index patients (128; 27; 242).
Grandpaternal inheritance of X-linked myotubular myopathy from a mosaic maternal grandfather has been described (104).
Genotype-phenotype correlations. X-linked myotubular myopathy predominantly arises from nonsense, frameshift, and splice site mutations in the MTM1 gene that typically result in loss of function. These pathogenic variations distribute across the gene without any specific concentration in particular domains (36; 131; 106; 25; 221). All males who carry pathogenic variants in the MTM1 gene will show symptoms of the disease. However, the severity of the illness can differ, ranging from mild to severe. In addition to small mutations, further reports present large deletions and duplications (24; 220; 05; 166; 189; 126; 88). Most point mutations are truncating, but approximately 30% are missense mutations that affect conserved residues (131; 21). Some of these are associated with a milder phenotype, although no strong correlation appears to exist between the nature of the mutation and the clinical picture, and severity varies even within families (131; 106; 155; 52; 204). In an Italian series of patients with autosomal and X-linked forms, the MTM1 gene was the most commonly mutated gene in the early-onset forms (74). In a few families, manifestations have been mild from the outset, and a 67-year-old grandfather was still mildly affected (12; 25; 240). An exceptional patient with progressive dementia had a mutation removing the start signal of exon 2 (154). In two studies, eighty-five percent of mothers were carriers, and approximately 5% of index cases were female (24; 119). The latter figure is, however, likely to be an underestimate because females with this X-linked disorder presenting as index cases may go undiagnosed (27; 242).
Pathogenesis. The emergence of pathological changes and distinct phenotypes in centronuclear myopathies can be attributed to the lack or insufficiency of proteins, including MTM1, BIN1 and DNM2. These proteins are functionally related to each other and are involved in autophagy, membrane fission, and vesicle trafficking (181). MTM1 plays a role in bringing BIN1 and DNM2 to the membranes of muscles, which helps enhance the formation of BIN1 tubules. Mutations in MTM1 affect the structure of the triad (182; 51). It has been shown that the proper balance of expression levels of BIN1, DNM2, and MTM1 is essential for muscle physiology. Furthermore, DNM2 appears to hold a significant position in the development of the condition and shows elevated levels in cases of centronuclear myopathy linked to BIN1, DNM2, and MTM1.
Myotubularins, a group of proteins associated with variety of diseases, have been found to be conserved throughout evolution and have similarities to phosphatases. Within the human system, there are 14 distinct paralogs of myotubularins; MTM1 absence being related to X-linked myotubular myopathy was the first one discovered and was followed by other myotubularin-related proteins, MTMR1 to MTMR13 (135; 172).
Mutations in two of these myotubularin-related genes, namely MTMR2 and MTMR13/SBF2, have been found to cause separate neuromuscular disorders: Charcot-Marie-Tooth disease type 4B1 (31; 18) and type 4B2 (193), respectively. Gomez-Oca and colleagues provide a review on the subject (89).
A boy with a big deletion involving both the MTM1 gene and the homologous MTMR1 gene did not differ clinically from other patients with myotubular myopathy in any significant way, indicating that the MTMR1 gene has little clinical significance during early postnatal life (241). Members of the myotubularin protein family form homodimers and also heterodimers in which an active and an inactive protein binding to each other may produce conformational changes affecting substrate affinity, hydrolysis, and binding to associated proteins (19; 123; 160; 47; 17).
MTM1 does not have the N-terminal extension (172). Interestingly, only a short version of MTMR2, which lacks this extension, shows a similar behavior to MTM1 in both yeast and mice. The production of MTMR2 isoforms using AAV vectors has been shown to improve myopathic characteristics in mice that lack MTM1 (56), suggesting that therapeutic interventions through MTMR2 may serve as a potential treatment for X-linked myotubular myopathy. Myotubularin is thought to be involved in the intracellular pathways necessary for myogenesis. Disease-causing mutations have been identified in all four active sites: GRAM, RID, PTP/DSP, and SID (106; 25; 21). These domains are implicated in substrate recognition, appropriate targeting to cellular compartments, and interaction with other proteins. Myotubularin was initially thought mainly to be a dual-specificity phosphatase acting on proteins but was later shown to be primarily a lipid phosphatase acting on phosphatidylinositol 3-monophosphate. It is involved in regulating intracellular and endosomal trafficking and vesicular transport processes (134; 53; 133; 30; 212; 132; 137; 222; 177; 41; 89). Gene expression analysis suggests that remodeling of the cytoskeletal and extracellular architecture may contribute to atrophy and disorganization of organelles in affected myofibers (164; 08).
A defect in muscle fiber maturation has been suggested as a pathogenetic mechanism in X-linked myotubular myopathy (196). However, studies have demonstrated the presence in muscle fibers of mature isoforms of myofibrillar proteins (194). A transgenic knockout mouse model confirmed these findings (38). This model also showed that in mice, myotubularin is necessary for the maintenance of skeletal muscle but not for myogenesis, including muscle fiber differentiation. A case report indicates that myofiber maintenance is the crucial problem in humans also (99).
Studies of a knockdown zebrafish model of myotubular myopathy confirmed the lipid phosphatase activity of myotubularin by showing high levels of its substrate phosphoinositide-3-phosphate in myotubularin-deficient muscle tissue (67). In other tissues, and in an experimental setting where myotubularin homologs were upregulated, the phosphatase function of myotubularin was found to be taken by homologous proteins. This provided an explanation for the specific involvement of muscle tissue in myotubular myopathy and suggested a possible route to future therapies.
The muscle tissue of the zebrafish model shows morphologic and functional abnormalities of the tubuloreticular system, paralleled by T-tubule abnormalities in patient muscle, suggesting that the pathogenetic mechanism in myotubular myopathy may be one of impaired excitation-contraction coupling. These results implicated a pathogenetic link not only to the other centronuclear myopathies (219; 182), but also to central core disease and multi-minicore disease. Impaired calcium homeostasis could, thus, be a possible common explanation for the known clinical and histologic similarities between these different congenital myopathies (67; 89; 140). These findings were further corroborated by studies on myotubularin-deficient fibers in the mouse model, showing fewer triads and ryanodine receptors than normal muscle fibers, as well as abnormal T-tubules and depressed calcium release from the sarcoplasmic reticulum (04). Altered calcium release has later been further corroborated as part of the pathogenetic pathway (130). Studies in human muscle suggest epigenetic modifications (10).
Myotubularin has been shown to be a desmin-binding protein with effects of the altered myotubularin on desmin-myotubularin interactions. Myotubularin deficiency also affects mitochondrial positioning, shape, dynamics, and function (108). Thus, an association of myotubularin was discovered with both sarcomeric thin filament abnormalities and mitochondrial abnormalities. Another study suggested that muscle atrophy may be induced by loss of myotubularin, leading to impaired Akt-dependent survival signaling (173). Apoptosis is also impaired (138).
Myotubularin phosphatases (eg, MTM1) play a crucial role in hydrolyzing cell membrane phospholipid phosphatidylinositol 3-phosphate (PI3P), which is necessary for autophagosome formation and endosome recycling to the cell surface to facilitate exocytosis in hepatocytes (122). Additionally, the accumulation of PI3P disrupts activity of phosphatidylinositol 3-kinase, an essential enzyme involved in the translocation of bile salt export pump (157). The inadequate activity of MTM1 within hepatocytes in patients with X-linked myotubular myopathy may impair the transportation of bile acids through the canalicular membrane. A direct link between liver abnormalities observed in X-linked myotubular myopathy and the MTM1 mutations has been suggested based on findings in a preclinical model. In this study, loss-of-function mutations in MTM1 led to notable liver abnormalities, such as disrupted bile flux and improper trafficking of transporters through endosomes in the zebrafish model (121). Additionally, the use of a DNM2 inhibitor was able to restore bile flow.
Studies towards treatment opportunities. Myotubularin appears to be required for the proper function of skeletal muscle in adulthood, which suggests long-term demands for the development of treatment (114).
Currently, Drosophila and zebrafish models provide further prospects for pathogenetic studies aiming toward identifying future therapeutic possibilities (118; 67; 96).
Myostatin has been proposed as a circulating biomarker in an Mtm1 mouse model (124). A study of the effects of inducing myostatin inhibition in myotubularin-deficient knock-out mice showed transiently better muscle strength and longer lifespan in the mice injected with the hypertrophy-inducing agent, ActRIIB-mfC, than in wild-type mice (141). The mice showed selective hypertrophy of type 2B fibers. A method for studying sarcomere structure and function in vivo in mice has been developed (156), and the proteomics of different mouse models have been studied (62).
Buj-Bello and coworkers reported successful viral transfer of myotubularin to selected murine muscles (37). Myotubularin replacement led to corrected positions of nuclei and mitochondria and to an increase in muscle volume as well as force. In the same report, results of overexpression of myotubularin in wild-type muscle indicated that myotubularin is involved in the plasma membrane homeostasis of muscle fibers.
This initial knockout mouse model, however, did not survive long enough for reliable preclinical testing of potential therapeutic modalities. A later knock-in mouse model with a longer lifespan and nonprogressive myopathy has been used for preclinical studies (170; 143; 148). Childers and colleagues described successful systemic AAV myotubularin gene delivery in both mice and dogs (46; 73). Further articles report improvement in MTM1-deficient mice by downregulation of dynamin 2 expression (50; 211; 45). Two trials of tamoxifen, previously used in mdx mice, in mouse models of X-linked myotubular myopathy showed promising results through the reduction of DNM2 and P13KC2B (84; 150). This exploration suggests the potential repurposing of tamoxifen for the treatment of individuals affected by X-linked myotubular myopathy. Currently, this potential repurposing is being studied in a clinical trial called TAM4MTM (NCT04915846).
AAV delivery of the mtm1 homolog mtmr2 also appears to have an ameliorating effect in mice (171; 56).
A study has presented initial evidence of epigenetic changes in X-linked myotubular myopathy (228). This abnormality, which was also observed in a mouse model, was effectively reversed by using a HDAC inhibitor.
Labrador, Rottweiler, and Boykin spaniel disorders resembling centronuclear myopathies in humans have been described, the natural course of the disease in dogs has been studied, outcome measures have been defined, and the canine models are being used for testing potential treatment modalities (216; 168; 118; 15; 93; 46; 86; 188; 87; 199; 200; 165). A 4-year follow-up study of dogs treated with gene therapy showed that the dogs are doing well; however, their muscle strength started to decline 3 years after the gene transfer (73; 71). AAV gene transfer appeared to correct fiber properties in these dogs (180).
In preparation for clinical trials, now ongoing, natural history studies have been reported (06; 16; 09; 92; 82). Of these, only one is truly prospective and longitudinal, albeit with a limited follow-up time of 1 year at the time of the first published report (09). The INCEPTUS natural history study focused on the findings of 34 children under the age of 4 years diagnosed with X-linked myotubular myopathy (66). Their median follow-up duration was reported as 13.0 months (ranging from 0.5 to 32.9 months). A paper suggested using the Bayesian statistical model for comparing each patient’s possible treatment response with the progression expected from their own natural history data (80). Outcome measures have been defined (70).
In many infants with this disorder, a strong fetal expression of vimentin and desmin has been found to persist in the muscle fibers of affected boys (186; 187; 196), but this is not a consistent feature. These intermediate filament cytoskeletal proteins attach to the membranes of the sarcolemma, nuclei, and mitochondria.
No reliable incidence figures are available. The disorder is rare; mutations have been reported in hundreds of patients from different parts of the world (229; 131; 106; 25; 21; 221; 24; 119). Many infants with this disorder may, however, have died undiagnosed whereas unusually mild cases may remain unidentified. A theoretical model has been described for estimating the prevalence (223).
Primary prevention is not currently feasible. Determination of carrier status and prenatal diagnosis is possible through mutation analysis (135; 134; 207; 208; 230; 21; 149). The frequency of new mutations appears to be rather low, and most mothers of affected boys are carriers of a mutated gene (134; 106; 21). A previous suggestion of genetic linkage heterogeneity for the X-chromosomal form of myotubular myopathy (185) was later invalidated (95). Mosaicism has been documented (227; 101) and needs to be taken into account in genetic counseling and prenatal diagnosis, but it remains to be determined how commonly it occurs (131).
Clinical manifestations are seen in a proportion of female carriers (232; 208; 100; 230; 205; 115; 128; 190; 94; 103; 74; 189; 27; 48; 174) and may be late (169). Muscle biopsy studies were previously used in carrier diagnosis (186; 32) but the carrier status of female relatives is best determined using direct mutation detection (135; 134; 207; 208; 149).
For surviving patients, a risk factor to keep in mind is any insidious respiratory difficulty (231; 198; 206). In some cases, this necessitates the use of a mechanical ventilator for intermittent or prolonged use (155). Other organs may become involved (105), and hepatic peliosis may ensue (98; 159). Treatment of peliosis has been done through arterial embolization (213) or liver transplantation (167). A follow-up study confirms that hepatobiliary involvement is common and unrelated to the severity of the muscle disorder (55).
Myotonic dystrophy in the neonate is clinically and histologically similar to X-linked myotubular myopathy, and myotonic dystrophy, being more common, is the more likely diagnosis in a female infant with this presentation. However, it has been recognized that female carriers of X-linked myotubular myopathy may manifest muscle disease, in at least a proportion due to skewed X-inactivation (208; 100; 230; 205; 115; 128; 190; 94; 169; 119; 189; 174). In a family, female carriers manifested spastic paraplegia (127). In all cases of X-linked myotubular myopathy in which a mutation has not been found in the MTM1 gene, it is necessary to exclude myotonic dystrophy. For this differential diagnosis, immunohistochemical testing for muscleblind-like 1 protein may be a more rapid method (195) than molecular genetic studies (34).
Profoundly affected boys with clinical features indistinguishable from those of X-linked myotubular myopathy have been diagnosed with RYR1-caused centronuclear myopathy (119).
Other disorders causing severe floppiness in combination with muscle weakness are the other congenital myopathies, severe childhood spinal muscular atrophy and other anterior horn cell disorders, myasthenia, and motor neuropathies. Prader-Willi syndrome with severe hypotonia and feeding difficulties can be excluded by molecular genetic methods.
In the case of a surviving male patient, it is important to differentiate between the X-linked and the autosomal forms of myotubular myopathy, the latter of which usually follow a milder course (232; 112). Four genes for autosomal forms have been characterized (29; 120; 162; 42), and muscle biopsy findings may provide clues for directing mutation detection (22; 118; 179). One workshop report provides a diagnostic algorithm for distinguishing between the various forms of centronuclear myopathy (118).
In many cases, there will be a family history of miscarriages or neonatal deaths of male infants in the maternal line. Pregnancy may have been complicated by polyhydramnios, and birth may have been premature. The floppy male infant is often long and light for his gestational age and has a large head. Spontaneous movements and respiration may be lacking. Ophthalmoplegia may be a feature but is not always apparent at birth.
Once clinical examination has shown muscle weakness and severe hypotonia not explained by abnormalities of the central nervous system, muscle biopsy is essential to confirm the diagnosis. Myotonic dystrophy may need to be excluded by molecular genetic methods, but other myopathies are excluded primarily by histopathologic differences in the muscle biopsy. Characteristic findings include centrally placed nuclei with a surrounding central zone deficient in oxidative enzyme activity, and some patients show so-called necklace fibers (22; 97).
Electromyography may yield normal findings or small polyphasic motor unit potentials, and sometimes even so-called neurogenic features. Serum concentrations of creatine kinase are usually normal, but sometimes they are slightly elevated.
The diagnosis of X-linked myotubular myopathy can be confirmed by direct molecular genetic testing of genomic DNA (135; 134; 207; 209; 230; 79; 106; 21; 74; 113), some cases requiring analyses of copy number variation (220; 24; 05; 166; 189; 126; 88). In a further subset of patients, RNA- or protein-based methods may be needed (178; 218; 226; 153; 02; 77; 35). In the absence of such confirmation, the autosomal forms should be considered (22; 118; 179; 119).
In all new families, a clinical geneticist should be involved in the workup, and the family should have access to genetic counseling. Muscle biopsy as a method for carrier detection has been replaced molecular genetic testing (135; 134; 207; 230; 21). It is to be noted that female carriers may show clinical symptoms, easily mistaken for manifestations of autosomal forms of the disorder (208; 100; 230; 205; 115; 128; 190; 103; 74; 189; 174). Carrier females with unilateral weakness have been described (68), and in one family, female patients had a clinical picture of hereditary spastic paraplegia (127). Of note, one affected boy with type 1 fiber predominance and unspecific myopathic changes was diagnosed only after the identification of a maternal relative with the same disorder (58).
At this time, no specific, curative treatment is commonly available, although therapy trials are underway. Nevertheless, much can be done for boys with X-linked myotubular myopathy (116; 206). No reliable prognostic indicators have been established (155), and active treatment of the neonate is indicated, at least initially, in view of the good outcome in some patients. It is recommended that any decision to refrain from active treatment should not be based on diagnosis alone but should be taken individually, using the same criteria as for children with other neonatally severe conditions (206).
For children who survive, management should be entrusted to a multidisciplinary team with experience of treatment of children with neuromuscular disorders. In particular, patients need regular evaluation of their respiratory capacity, and they are likely to benefit from early intervention including intermittent use of a mechanical ventilator (231; 234; 198; 206). Based on studies of four essentially nonambulant patients, Cahill and colleagues make recommendations for the orthopedic treatment of scoliosis by long posterior fusion, and of metaphyseal fractures of long bones by brief periods of immobilization, while suggesting conservative treatment for contractures (40).
Increasing evidence indicates that hepatobiliary issues are linked to the natural progression of X-linked myotubular myopathy and are unrelated to any particular treatments. The evidence emphasizes the significance of monitoring liver parameters, potentially consulting a pediatric hepatologist, as a crucial facet of ongoing clinical care. Assessing liver-related laboratory values, such as ALT, AST, GGT, and total and direct bilirubin, alongside examining serum bile acid levels, could aid in detecting cholestatic episodes among individuals with X-linked myotubular myopathy (66). The families should be provided with access to genetic counseling.
Proof of concept has been achieved for several different therapy modalities using replacement of MTM1 through viral transfer of a normal gene or a homolog, enzyme replacement therapy, reduction of the interacting protein dynamin or drugs acting on the neuromuscular junction or the use of tamoxifen, or modification of the autophagy pathway. Several studies provide reviews on the subject (89; 140; 85). This brightens the outlook for future therapeutic trials, and preparations are in progress, covering many aspects of the disorder (37; 176; 03; 75; 139; 46; 50; 61; 86; 129; 183; 73; 172; 84; 150; 56; 210; 180; 62; 70; 175). The first human trial, ASPIRO, testing resamirigene bilparvovec as AAV-mediated gene therapy for X-linked myotubular myopathy, took the form of an open-label dose-escalation study (197). Within this trial, 17 patients with X-linked myotubular myopathy were allocated to the higher dose group, seven patients to the lower dose group, and 14 individuals to a control group. The median ages at the time of dosing or enrollment were recorded as 12.1, 31.1, and 18.7 months for the lower dose, higher dose, and control groups, respectively. The study documented improvements in both ventilatory capacity and motor function. However, the deaths of four participants imposed a clinical hold on the trial in September 2021; this hold persisted as of December 2023. Among the four participants who were administered doses and later deceased (three receiving the higher dose and one receiving the lower dose), each experienced severe cholestatic liver injury and decompensated liver failure. Additionally, five participants experienced nonfatal, treatment-associated serious adverse events related to the hepatobiliary system. Myocarditis is another concern that has emerged in some patients after treatment with resamirigene bilparvovec.
Although histopathological endpoints, such as fiber type distributions, numbers of triad structures, the degree of internal/central nucleation, fiber type distributions, and numbers of satellite cells, did not demonstrate statistically significant changes following treatment with resamirigene bilparvovec, muscle biopsies taken at 24 weeks post-treatment showed marked improvements in organelle localization, without apparent increases in myofiber size among most participants (142). However, statistically significant increases in myofiber size were detected in nine biopsies obtained at 48 weeks.
The economic disease burden of X-linked myotubular myopathy has been studied in the United States (184), but the results may not be applicable to other countries as both health economy and treatment policies vary between countries (155).
Pregnancies in which the fetus has X-linked myotubular myopathy are often complicated by polyhydramnios, and premature births are common. Continued pregnancies require careful follow-up. Planning for the delivery should be a collaborative effort between the pregnant woman, her obstetrician, and the anesthetist, and a pediatrician should be on call at delivery.
Although malignant hyperthermia is not recognized as a specific risk factor in the anesthesia of patients with X-linked myotubular myopathy, the anesthesiologist needs to be informed of the patient's diagnosis (90; 33; 49). Muscle relaxants are probably best avoided, and benzodiazepines should be used with caution because of their potentially adverse effects on breathing and muscle power. Careful evaluation of respiratory capacity should be performed before administering anesthesia (231; 234; 198). In especially severe cases, positioning during anesthesia may require special attention (78).
All contributors' financial relationships have been reviewed and mitigated to ensure that this and every other article is free from commercial bias.

Haluk Topaloglu MD
Dr. Topaloglu of Hacettepe Children's Hospital in Ankara, Turkey, has no relevant financial relationships to disclose.
See Profile
Gulcin Akinci MD
Dr. Akinci of Izmir Faculty of Medicine and Dr. Behcet Uz Children's Hospital has no relevant financial relationships to disclose.
See Profile
Harvey B Sarnat MS MD FRCPC
Dr. Sarnat of the University of Calgary has no relevant financial relationships to disclose.
See ProfileNearly 3,000 illustrations, including video clips of neurologic disorders.
Every article is reviewed by our esteemed Editorial Board for accuracy and currency.
Full spectrum of neurology in 1,200 comprehensive articles.
Listen to MedLink on the go with Audio versions of each article.
MedLink®, LLC
3525 Del Mar Heights Rd, Ste 304
San Diego, CA 92130-2122
Toll Free (U.S. + Canada): 800-452-2400
US Number: +1-619-640-4660
Support: service@medlink.com
Editor: editor@medlink.com
ISSN: 2831-9125
Neuro-Oncology
Jan. 14, 2025
Neuromuscular Disorders
Dec. 29, 2024
Developmental Malformations
Dec. 26, 2024
Developmental Malformations
Dec. 26, 2024
General Child Neurology
Dec. 26, 2024
Developmental Malformations
Dec. 14, 2024
Developmental Malformations
Dec. 12, 2024
Neuromuscular Disorders
Dec. 09, 2024